Article Text

Statistics from Altmetric.com
Sudden death in childhood is rare. About 10% of paediatric deaths after the first year of life are sudden and population based studies put the individual age related risk at around 1:20 000 to 1:50 000 per year.1–4w1–3 About half of these deaths are related to a previously known abnormality, the most common being epilepsy, asthma, and cardiovascular abnormalities. Another third are attributed to an abnormality discovered at necropsy, usually either an infection or a cardiovascular abnormality. At least one sudden death in six remains unexplained, but this is almost certainly an underestimate as some deaths attributed by the coroner's pathologist to epilepsy or respiratory infection are probably more accurately described as being unexplained by findings at necropsy.4w4
MECHANISMS OF SUDDEN DEATH
Although all deaths result in asystole, not all sudden deaths are caused by arrhythmias. The precise mechanism of sudden death depends upon the cause. One report of terminal electrical activity in paediatric patients dying in hospital documented bradycardic arrest in 88% of neonates, 67% of infants, and 64% of children.5 Ventricular tachycardia or fibrillation was more likely in those with heart disease and in older children. The term “sudden death” should not be confused with non-fatal cardiac arrest.w5
Sudden cardiac death in infancy
Sudden death in infancy is usually caused either by infection or by sudden infant death syndrome. A few neonatal or infant deaths are caused by unrecognised congenital cardiovascular malformations, particularly duct dependent abnormalities or obstructive left heart malformations.w6 Primary arrhythmias are rare causes of death in infancy but fatal ventricular arrhythmias are described.6 Complete atrioventricular block is usually recognised in utero or soon after birth but may cause death if unrecognised or untreated.w7
Sudden death in children with postoperative congenital heart disease
In the 1960s and 1970s sudden cardiac death most often occurred in children with irreversible pulmonary vascular disease associated with unoperated congenital heart disease or in children with unoperated aortic valve stenosis.w8 w9 In recent years surgical repair has been performed earlier and more effectively so that those most at risk of sudden death now are children with repaired heart disease. In a population based study of late postoperative sudden death, Silka and colleagues identified an average risk of 0.9 per 1000 patient-years follow up for the most common surgically repaired malformations.7 Those patients with a risk above the average had aortic valve stenosis, transposition of the great arteries, tetralogy of Fallot or coarctation of the aorta. Death was attributed to “arrhythmia” in the majority, based on the history, but in only a few was an arrhythmia identified in life.
Sudden death in adults with congenital heart disease
Among patients in an adult congenital heart follow up clinic in Toronto, not all of whom had undergone surgery, the reported sudden death rate was 5.3 per 1000 patient-years.8 The most common abnormalities in those who died suddenly were Eisenmenger's syndrome, tetralogy of Fallot, and transposition of the great arteries, but the risk for individual diagnoses could not be assessed for lack of a denominator. In a more recent report from the same unit, 8% of adult patients died during follow up—65% of deaths were cardiovascular and 26% were sudden.w10 Although numbers for individual diagnoses were small, the highest proportion of deaths were sudden in patients with coarctation of the aorta, Ebstein's anomaly, and congenitally corrected transposition of the great arteries. The highest number of sudden deaths in the clinic population occurred in patients who had undergone repair of tetralogy of Fallot.
Sudden death after repair of tetralogy of Fallot
Of the various problems encountered late after surgical repair of tetralogy of Fallot, sudden death is the most difficult to predict. It usually occurs many years after operationw11 and thus affects young adults more than children, with an average risk of 1.4 per 1000 patient years, or about 1 in 700 per year.9 Many risk factors for sudden death have been identified retrospectively, but prospectively, even in combination, they are not useful in predicting risk.10w12 w13 Although ventricular tachycardia or fibrillation is thought to be the most common mechanism of sudden death, more minor ventricular arrhythmias are so common as to be unhelpful in predicting risk. An important recent “pseudo-prospective” multicentre study involving six centres in the UK, USA, Canada, and Japan retrospectively identified 793 patients alive in 1985 and “followed” their progress for the next 10 years, yielding more than 7500 patient-years follow up.11 Thirty three patients developed sustained monomorphic ventricular tachycardia, 29 developed atrial flutter or fibrillation, 16 died suddenly, and 715 remained free from arrhythmia. There were no deaths in those who presented with ventricular tachycardia and there was only one death among patients presenting with atrial arrhythmia. The risk profile for ventricular tachycardia and sudden death was similar, with most of the patients having pulmonary regurgitation and right ventricular dysfunction. The authors suggest that surgical preservation or restoration of pulmonary valve function may reduce the risk of sudden death, but this remains unproven.
Sudden death after atrial repair of transposition of the great arteries
Most major published reports of experience of the Senning and Mustard operations for atrial repair of transposition of the great arteries give a risk of 5–6 per 1000 patient-years or of about 1 in 180 per year.9 The relation between late sudden death and arrhythmia is not clear. There is a progressive loss of sinus rhythm so that fewer than half of patients have normal rhythm after 10 years.w14 Junctional rhythm is common but asymptomatic bradycardia does not require treatment. Bradycardia seems not to be a risk factor for sudden death and pacemaker implantation offers no protection against it. One widely accepted theory is that the sudden onset of atrial flutter with 1:1 atrioventricular conduction may lead to sudden death, particularly if it is associated with impaired right ventricular function or atrial baffle obstruction, but as yet there is only circumstantial evidence for this.w15 There is a complicated interrelation between arrhythmia, impaired ventricular function, and sudden death, which has yet to be explored fully.w16 Other possible causes for late death after atrial repair of transposition include ventricular arrhythmias7 or acute heart failure.
Sudden death after repair of other malformations
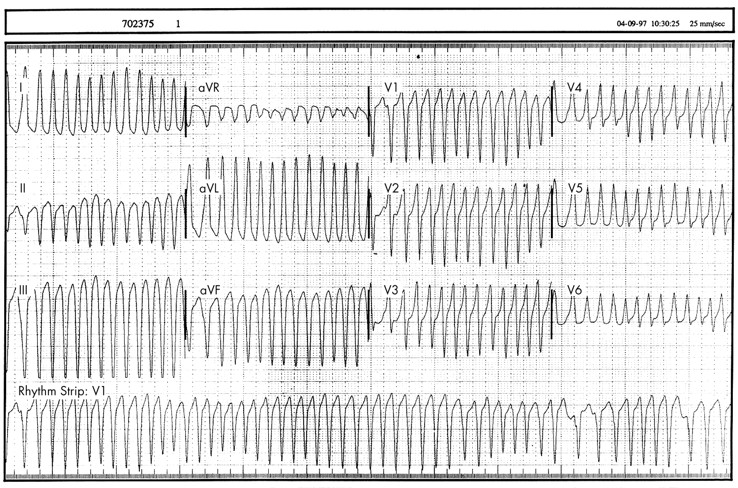
Late sudden death after surgical repair of other common cardiovascular malformations is rare, with an incidence of around 0.1 per 1000 patient-years.7 Surgical repair of some less common cardiac malformations may be associated with a higher risk of late sudden death. In a report of experience with the Rastelli operation for repair of complex transposition of the great arteries from Boston, there were five late sudden deaths in a group of 94 survivors of surgery who were followed for a median of 8.5 years (6.3 deaths per 1000 patient years).w17 Deaths were thought to be caused by the development of ventricular arrhythmias (fig 1) or atrioventricular block. The late postoperative sudden death rate in patients with heart defects characterised by double outlet right ventricle may be even higher.w18 Among 89 patients the sudden death rate was 26 per 1000 patient years, with 50% of deaths within one year of surgery. Risk factors for late death included perioperative ventricular tachycardia and atrioventricular block. The risk in rare conditions is harder to define but in some, such as pulmonary atresia, it may be significant.w19

Postoperative ventricular tachycardia in a 14 year old boy with a Rastelli repair of complex transposition who presented with syncope. The left bundle branch block pattern and inferior frontal plane vector indicate an origin from the right ventricular outflow. After electrophysiology study he underwent defibrillator implantation.
Sudden death in children with unoperated heart disease
One would hope that most significant cardiovascular malformations would be detected early in life, preferably at an asymptomatic stage. However, despite health screening some significant problems go unrecognised,w20 and may cause sudden death. Deaths from obstructive left heart malformations are unusual but they do occur.4 Postoperative deaths in such patients were ascribed to “arrhythmia” in the study by Silka and colleagues, but syncope in aortic valve stenosis is probably not primarily arrhythmic in origin.w21 Congenital abnormalities of the coronary arteries are rarely diagnosed in life and prodromal syncope is uncommon. The most frequently recognised abnormalities after sudden death are anomalous origin of the left or right coronary arteries from the contralateral aortic sinus, with a proximal course between the aorta and the main pulmonary artery.w22 w23 Although the prevalence of such anomalies is hard to define precisely, the risk of sudden death is probably high enough to warrant surgical treatment.w24 Sudden death in other specific conditions, such as Williams syndrome, is also reported and may be caused, in some cases at least, by coronary artery stenosis or malformation.w25
Sudden death in hypertrophic cardiomyopathy
The risk of sudden death in childhood from hypertrophic cardiomyopathy diagnosed in life is impossible to establish for lack of a denominator. Hypertrophic cardiomyopathy is said to affect as many as 1 in 500 of the adult population, but it is much less common in childhood. The impression given by published reports from specialised referral units is that the risk of sudden death is high whereas evidence from the few population based studies does not support this.12w26 w27 “Risk stratification” is encouraged and may identify a few individuals at increased risk.13w28 “Risk factors” have been identified but are absent in most children. Risk factors for an increased risk of sudden death in adults include a family history of sudden death from hypertrophic cardiomyopathy, recurrent syncope, non-sustained ventricular tachycardia on an ambulatory ECG, and extreme left ventricular hypertrophy, but their presence does not reliably identify individuals at risk. A recent study of the use of implantable cardioverter-defibrillators (ICDs) in primary and secondary prevention has shown benefit.w29 Appropriate discharges were triggered by ventricular tachycardia or fibrillation, providing insight into the mechanism of sudden death and an incentive for better risk stratification. However, only eight of 128 patients receiving an ICD were children. In a study of 99 children with hypertrophic cardiomyopathy in Toronto, the reported annual combined risk of sudden death and cardiac arrest was 2.7% between the ages of 8 and 18 years.w30 In this study syncope did not predict sudden death whereas cardiac arrest did and ventricular tachycardia on ambulatory ECG was a significant risk factor. The risk of sudden death is low for the majority of children, especially in the absence of symptoms. There is no evidence that treatment of asymptomatic patients has any effect on outcome.w31 Because some sudden deaths occur during or after exercise it is often recommended that affected children should avoid sports or intense physical exertion. While such advice may seem sensible, there is no evidence that this leads to any reduction in risk.12
Sudden death from previously unsuspected hypertrophic cardiomyopathy is rare—with an age specific risk in apparently normal children or adolescents of less than 1 in 1 000 000 per year.4w32 Because hypertrophic cardiomyopathy typically has dominant inheritance, diagnosis at necropsy should lead to screening of other family members. Often no further cases are identified, either because of variable penetrance or because the index case represents a new mutation. Clinical exclusion of the diagnosis is often very difficult as detectable cardiovascular abnormalities may only develop during adolescence.13 Gene testing is not generally available at present.14 A non-hypertrophic variant of hypertrophic cardiomyopathy has also been described recently.w33 Troponin T mutations produce characteristic histological myocardial disarray and seem to have a particularly poor prognosis in the few families described but their prevalence is unknown.
Main causes of sudden cardiac death in children
Cardiovascular malformations
-
▸ Atrial repair of transposition of the great arteries (Mustard and Senning operations)
-
▸ Repaired tetralogy of Fallot
-
▸ Aortic valve stenosis
-
▸ Coarctation of the aorta
-
▸ Pulmonary atresia
-
▸ Pulmonary vascular disease (Eisenmenger syndrome)
-
▸ Coronary artery malformations
Other structural heart abnormalities
-
▸ Hypertrophic cardiomyopathy
-
▸ Myocarditis
-
▸ Dilated cardiomyopathy
-
▸ Kawasaki disease
-
▸ Arrhythmogenic right ventricular cardiomyopathy
Primary arrhythmias
-
▸ Long QT syndrome
-
▸ Wolff-Parkinson-White syndrome
-
▸ Atrioventricular block
-
▸ Other ventricular arrhythmia (for example, catecholaminergic ventricular tachycardia, Brugada syndrome, etc)
-
▸ Non-hypertrophic varieties of hypertrophic cardiomyopathy (troponin T mutation)
Sudden death in other cardiomyopathy
Sudden death in dilated cardiomyopathy is uncommon. A study of 63 children from Houston documented 16% mortality over 10 years, but only one of five deaths was sudden.w34 In a report from Heidelberg four of 28 children died suddenly during a mean follow up of 4.1 years.w35 In both studies arrhythmias of various types were present in at least half of the children, but they were unhelpful in risk prediction.
Restrictive cardiomyopathy is a rare diagnosis at any age. There is a high mortality in childhood with a two year survival of only 50%.w36 Most deaths are sudden. The main predictors of mortality are syncope, chest pain, ischaemic changes on the ECG, and female sex. Cardiac transplantation is the only treatment to offer an improvement in outcome.w36
Arrhythmogenic right ventricular cardiomyopathy is a familial disease which usually affects young adult men.w37 True diagnoses in childhood are exceptional. The disease may present with syncope, or with ventricular tachycardia with left bundle branch block morphology (that is, right ventricular origin). It may cause death in young athletes and seems to be particularly common in Italy.w37 Risk assessment and therapeutic strategies are not well developed.15
The incidence of sudden death in apparently normal children beyond the first year of life is probably around 1–1.5 per 100 000 per year.4 In addition to the various types of cardiomyopathy and coronary artery abnormalities discussed above, diagnoses first established after death include myocarditis.4,16w38 The exact mode of death in myocarditis is uncertain but it is probably caused by an arrhythmia. In other children death may remain unexplained after necropsy, but the circumstances may lead to a retrospective presumed diagnosis of a primary cardiac arrhythmia.
SUDDEN DEATH IN CHILDREN WITH PRIMARY CARDIAC ARRHYTHMIAS
Most sudden cardiac deaths that remain unexplained after necropsy are probably caused by primary cardiac arrhythmias. Arrhythmias which are known to be potentially fatal, and which would leave no trace after death, include polymorphic ventricular tachycardia in congenital long QT syndrome,17w39 other primary ventricular arrhythmias such as those described by Brugadaw40 w41 and Coumel,w42 atrial fibrillation in Wolff-Parkinson-White syndrome,w43 and congenital complete atrioventricular block. Retrospective confirmation of an arrhythmia is not possible unless a familial condition (such as long QT syndrome) is subsequently recognised in a family member. This group of arrhythmias also comprises the majority of diagnoses made after resuscitation from out of hospital cardiac arrest.w44 w45
Sudden death in congenital long QT syndrome
Congenital long QT syndrome is characterised by QT prolongation on the ECG in association with polymorphic ventricular tachycardia, and may affect as many as one child in 7000.6 It is usually amenable to treatment with a β blocker, but some patients require pacemaker or defibrillator implantation.w46 It is difficult to establish the overall mortality risk as death may be the first symptom, but 8% of 287 patients reported by the Pediatric Electrophysiology Society died suddenly during a mean follow up of five years.17 The main predictors of sudden death in that report were extreme QTc prolongation (> 600 ms) and poor compliance with β blocker medication. Congenital long QT syndrome has dominant inheritance. Despite this, in the Pediatric Electrophysiology Society report only 39% of children with a clinical diagnosis had a positive family history of long QT syndrome, probably explained either by variable penetrance or because they represent new mutations. Diagnosis in a child should lead to screening of first degree relatives. However, long QT syndrome can be difficult to diagnose or to exclude because of the overlap of QT intervals in gene carriers and in the normal population. Although several gene defects causing long QT syndrome have been identified, genetic analysis is costly and time consuming and the diagnosis remains clinical in most cases. Because a normal phenotype in the presence of an abnormal genotype is associated with a risk of syncope or death, there may be an indication for genetic investigation of family members with apparently normal or borderline ECGs.14
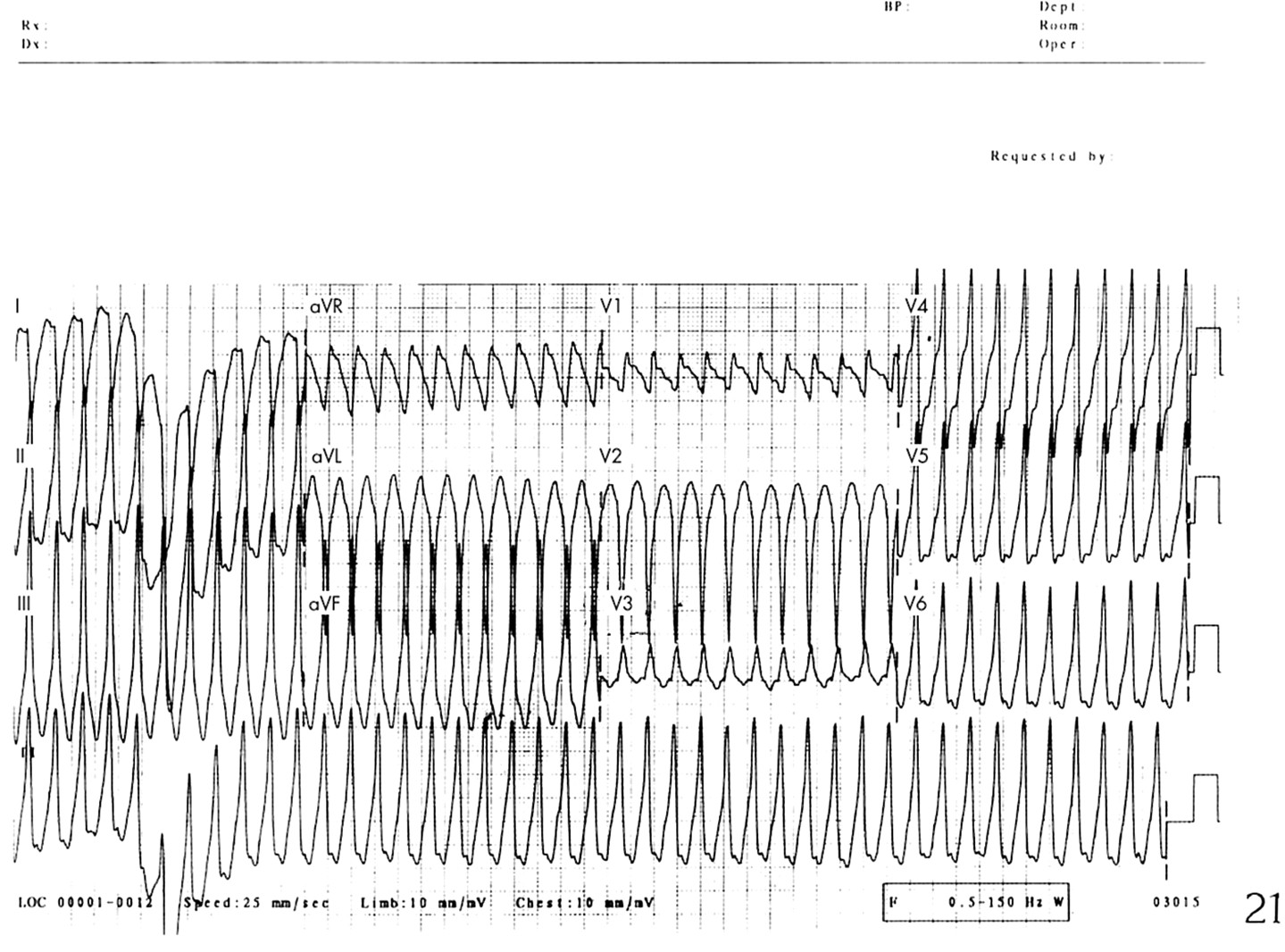
Acquired long QT syndrome is the term sometimes used to describe QT prolongation seen in metabolic disturbances or with use of pharmacological agents. It may also be familial and may cause arrhythmias and sudden death (fig 2).w46

Sudden death in acquired QT prolongation. The ambulatory recording shows development of polymorphic ventricular tachycardia which degenerates into ventricular fibrillation and then asystole.
Sudden death in other ventricular arrhythmias
Catecholaminergic ventricular tachycardia, as described by Coumel, has some similarities with long QT syndrome.w42 However, the QT interval is normal and polymorphic ventricular tachycardia is preceded by increasingly complex ventricular arrhythmias, including a characteristic bi-directional ventricular tachycardia. The exercise test and ambulatory ECG are often positive and are helpful in making the diagnosis. Treatment with a β blocker is usually effective but poor compliance with medication is associated with a high risk of death.
Brugada syndrome describes the association of polymorphic ventricular tachycardia or ventricular fibrillation with ST segment elevation in right precordial leads on the ECG.w40 41 It is a familial condition, usually not causing symptoms until early adult life, and is probably very rare in children. Diagnosis is difficult as the characteristic ECG changes are intermittent and can be very subtle. The only means of preventing sudden death in affected individuals is an implantable defibrillator.
Sudden death in Wolff-Parkinson-White syndrome
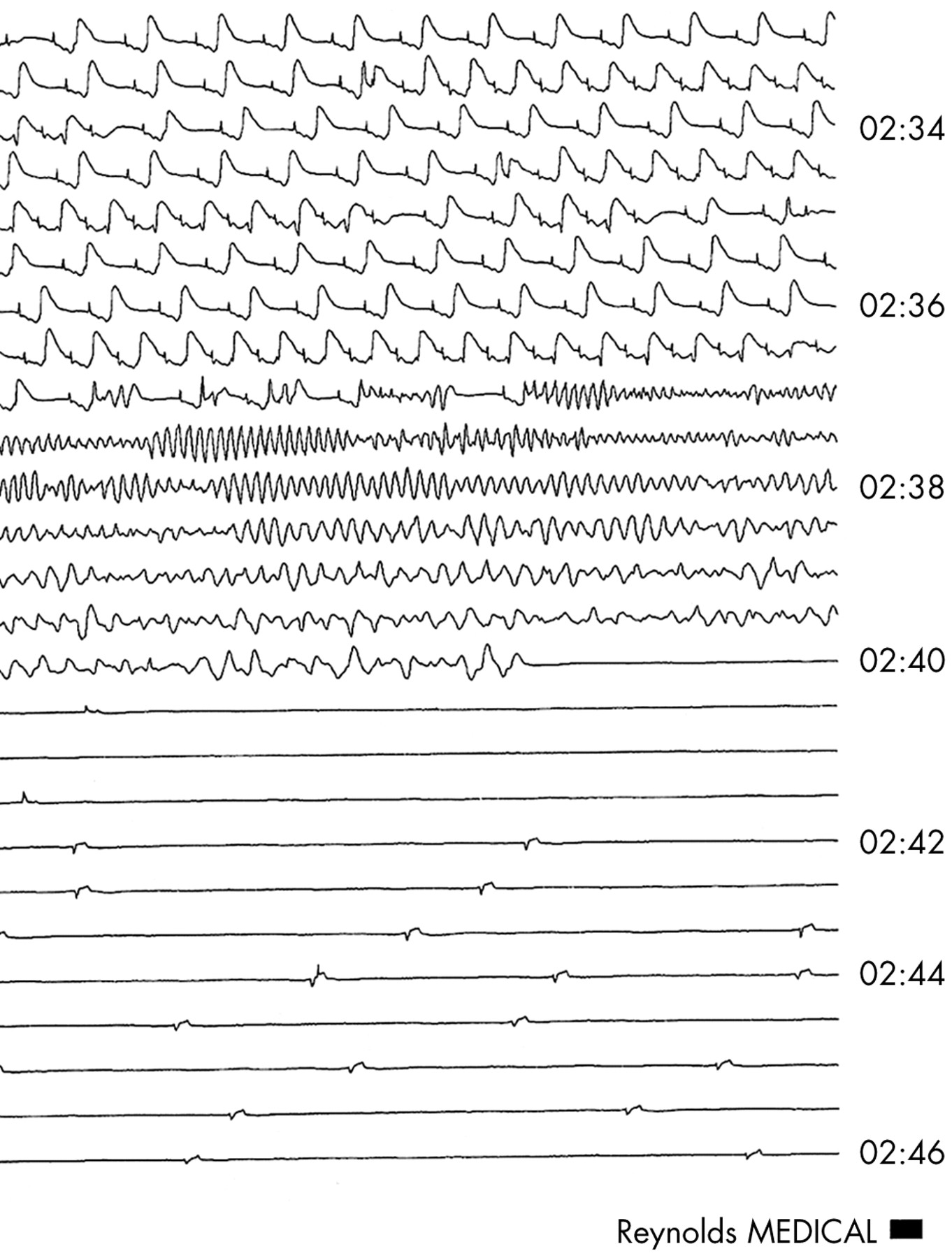
Wolff-Parkinson-White syndrome is one of the most common causes of arrhythmias in childhood, but sudden deaths are rare. The overall mortality risk is very difficult to quantify but may be around 1.5 per 1000 patient-years.w47 Sudden death is probably caused by ventricular fibrillation which, in turn, is precipitated by atrial fibrillation in patients with a very short anterograde accessory pathway refractory period (fig 3). Cardiac arrest and sudden death mainly occur in adolescents and young adults and are very rare in younger children.w48 Resuscitation from cardiac arrest is (perhaps not surprisingly) a strong predictor of risk, but syncope and atrial fibrillation have poor predictive value, although they would be indications for catheter ablation in any case. Although a short anterograde refractory period of the accessory pathway is thought to be the best electrophysiological indicator of risk, the predictive value of such a finding is poor.w48 Patients with significant symptoms require treatment, irrespective of any associated small risk, but the role of risk stratification by invasive electrophysiology study in children with few or no symptoms remains unclear.
{kind=link}
{kind=link}
{kind=link}
Atrial fibrillation in Wolff-Parkinson-White syndrome in a 10 year old girl with a history of recurrent syncope. There is a rapid, irregular, wide QRS tachycardia with a morphology which matched the pre-excitation seen in sinus rhythm. The accessory pathway was ablated.
Sudden death in congenital atrioventricular block
Congenital complete atrioventricular block affects around one baby in every 20 000. Not all are recognised at birth, or even in infancy.w7 The risk of sudden death is probably mainly related to the ventricular rate. The development of symptoms is an indication for pacemaker implantation, but absence of symptoms in childhood or adult life does not imply absence of risk of sudden death.w7 w49 Risk stratification in asymptomatic patients is imperfect but pacemaker implantation is usually advisable in infants with a resting ventricular rate below 55 per minute, or in children below 50 per minute, especially if there are other arrhythmias or other findings on ambulatory monitoring.w50
SUDDEN DEATH IN YOUNG ATHLETES
Deaths during sports attract publicity but they are rare events. Estimates from the USA suggest that 5 per 100 000 young athletes (including children, teenagers, and young adults) have a predisposing condition and that about 10–25 such deaths per year occur in the US population of 260 000 000.18 The most common structural abnormalities identified at necropsy are hypertrophic cardiomyopathy, coronary artery abnormalities, right ventricular cardiomyopathy, and aortic valve stenosis.16 “Unexplained” cases may be caused by ventricular arrhythmia or atrial fibrillation in Wolff-Parkinson-White syndrome (as discussed above). Health screening programmes are in place for some young athletes but their efficacy has not been prospectively evaluated.
SYNCOPE IN APPARENTLY NORMAL CHILDREN
Syncope is a frequent problem in childhood. The common causes are benign whereas some rare causes are potentially dangerous.19 A “simple faint”, or vasovagal syncope, is said to occur in up to 15% of the normal population at some time during childhood. Other “vascular” but benign causes of syncope include neurocardiogenic syncope in older children and “pallid syncope” or “reflex asystolic syncope” in younger children. The mechanisms of these may be similar with inappropriate reflex bradycardia and hypotension.w51 In the former syncope is often postural and in the latter a frequent trigger is surprise, minor hurt or frustration. Children or adults with repaired or unoperated congenital heart disease and children with other known heart disease should already be under review in a specialist follow up clinic. Syncope in such patients may be an indication for urgent reassessment or investigation.
Possible ECG abnormalities in children with syncope
-
▸ QT prolongation
-
▸ Ventricular pre-excitation
-
▸ Atrioventricular block
-
▸ Left ventricular hypertrophy or ST/T abnormalities in hypertrophic cardiomyopathy
-
▸ ST elevation in V1–V3 in Brugada syndrome
Studies that examine children presenting with syncope rarely identify any diagnoses with a significant risk of death because such problems are rare. However, the spectrum of underlying abnormalities in children resuscitated from cardiac arrest is, not surprisingly, very different and more malignant. Diagnoses include various types of cardiomyopathy and substrates for primary arrhythmias including QT prolongation and ventricular pre-excitation, all discussed above.w44 w45
Investigation of syncope
The history is of paramount importance in the assessment of children with syncope. Common but benign causes such as vasovagal syncope or hyperventilation are usually easily recognised. A history of syncope during or immediately after exertion, syncope preceded by palpitations, or syncope in the presence of a family history of premature sudden death, congenital long QT syndrome or hypertrophic cardiomyopathy should lead to referral for specialist evaluation. Detection of a cardiac murmur (or other abnormal signs on examination) should also lead to specialist referral as it may be a clue to the presence of aortic valve stenosis, coarctation of the aorta or hypertrophic cardiomyopathy.
The ECG is a valuable screening test in children with syncope and may identify QT prolongation, ventricular pre-excitation or atrioventricular block, or may be a clue to the presence of underlying ventricular arrhythmia or cardiomyopathy. Other investigations may include echocardiography, ambulatory or exercise electrocardiography or invasive electrophysiology, but the investigative strategy depends fundamentally on the history.
Sudden death in children and adolescents: key points
-
▸ Sudden death accounts for about 10% of all deaths in children
-
▸ Structural cardiac abnormalities that may cause sudden death in apparently normal children include hypertrophic cardiomyopathy, other cardiomyopathies, and coronary artery malformations
-
▸ The operations with the most significant risk of late sudden death are the Senning or Mustard operations for transposition of the great arteries and repair of tetralogy of Fallot
-
▸ At least one sudden death in six remains unexplained—an age specific incidence of around 1 in 200 000 per year
-
▸ Deaths which remain unexplained after necropsy are probably caused by primary cardiac arrhythmias, such as those in long QT syndrome, other primary ventricular arrhythmias, Wolff-Parkinson-White syndrome, and congenital atrioventricular block
POPULATION SCREENING FOR UNSUSPECTED HEART DISEASE
The infrequency of prodromal symptoms and the absence of physical signs in children who die suddenly and are found at necropsy to have a cardiac problem may seem to be an indication for screening of the general population. However, there is no evidence that population screening of children for abnormalities such as hypertrophic cardiomyopathy is either feasible or appropriate, and no evidence that either early detection or treatment in the absence of symptoms has any effect on outcome. The only prospective population wide screening programme of normal children has been undertaken in Tokyo.20 The reported prevalence of hypertrophic cardiomyopathy is around 1 in 15 000 and those children identified seemed to be at particularly low risk.w52 The programme does also detect children with long QT syndrome, atrioventricular block, and Wolff-Parkinson-White syndrome by routine ECG but these problems are rare. So far it has not been possible to show that screening prevents sudden death.
SUMMARY
Many children who suffer sudden cardiac death are already known to have a heart problem. Many of these have had surgery for congenital heart disease and the risk of sudden death in such patients probably increases with time. The risks for such patients are probably decreasing with better and earlier surgical repair, but prospective identification of individuals at significant risk remains an elusive goal. In some children who die suddenly the necropsy reveals the cause of death. In others, who are apparently normal, the likely cause is a primary cardiac arrhythmia. Only a minority of children who die suddenly have previously experienced syncope.
Most children who experience syncope are not at risk of sudden death. Worrying features which would be an indication for investigation include syncope during or immediately after exercise, collapse in a swimming pool, a family history of premature sudden death or of potentially life-threatening cardiac problems, and abnormalities on clinical examination or ECG.
Several potentially fatal conditions are genetic in origin but unfortunately, at present, many of them are easier to confirm than to exclude. For instance, an echocardiogram during childhood cannot rule out the possibility of later manifestation of hypertrophic cardiomyopathy; some gene carriers of long QT syndrome have normal ECGs and yet 2.5% of normal children have a QT interval above the “upper limit of normal”; and the characteristic ECG appearances of Brugada syndrome are intermittent. For the moment identification of these conditions remains a clinical problem but in the future they will probably be simple genetic diagnoses. Some of the clinical management dilemmas, however, will probably remain.
REFERENCES
Supplementary materials
Congenital heart disease
SUDDEN DEATH IN CHILDREN AND ADOLESCENTS
Christopher Wren
Web-only References
[View PDF]
Linked Articles
- Miscellanea