Article Text

Abstract
Background High blood pressure (BP) during acute stroke is associated with poorer stroke outcome. Trials of treatments to lower BP have not resulted in improved outcome, but this may be because treatment commenced too late. Emergency medical service staff (paramedics) are uniquely placed to administer early treatment; however, experience of prehospital randomised controlled trials (RCTs) is very limited.
Methods We conducted a pilot RCT to determine the feasibility of a definitive prehospital BP-lowering RCT in acute stroke. Paramedics were trained to identify, consent and deliver a first dose of lisinopril or placebo to adults with suspected stroke and hypertension while responding to the emergency call. Further treatment continued in hospital. Study eligibility, recruitment rate, completeness of receipt of study medication and clinical data (eg, BP) were collected to inform the design of a definitive RCT.
Results In 14 months, 14 participants (median age=73 years, median National Institute of Health Stroke Scale=4) were recruited and received the prehospital dose of medication. Median time from stroke onset (as assessed by paramedic) to treatment was 70 min. Four participants completed 7 days of study treatment. Of ambulance transported suspected stroke patients, 1% were both study eligible and attended by a PIL-FAST paramedic.
Conclusions It is possible to conduct a paramedic initiated double-blind RCT of a treatment for acute stroke. However, to perform a definitive RCT in a reasonable timescale, a large number of trained paramedics across several ambulance services would be needed to recruit the number of patients likely to be required.
Clinical trial registration http://www.clinicaltrials.gov. Unique identifier: NCT01066572.
- Paramedics, Clinical Management
- Prehospital Care, Clinical Management
- Research, Clinical
- Stroke
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
Hypertension in acute stroke is associated with poor functional outcome, but its optimal treatment immediately after stroke is unclear.1 Studies that have lowered blood pressure (BP) soon after stroke have not shown improved outcome,1–3 but this may be because treatment commenced too late. Rapid neurological injury occurs following stroke and previous BP-lowering trials have enrolled patients several hours to days after stroke symptom onset.1 The earliest that BP-lowering treatment could commence is during contact with ambulance paramedics.
While paramedics are uniquely placed to deliver early treatments, their experience of participating in randomised controlled trials (RCTs) to evaluate interventions is limited and few trials have been conducted within ambulance services.4 ,5 This is likely to be because prehospital research infrastructure is still in development6 and because the logistics of RCTs are more challenging in this environment.7 ,8 In terms of prehospital stroke trials, experience is very limited with only one US conducted pilot RCT evaluating pre-hospital treatment with magnesium,9 from which a definitive study is now underway (FAST-MAG).10
When the feasibility of a RCT is unclear, it is important to conduct an external (rehearsal) pilot trial to inform the design of a definitive study.11 ,12 We report the results of a UK conducted pilot double-blind RCT of paramedic initiated lisinopril for treatment of hypertension in acute stroke.
Methods
Study design and setting
The study design has been reported in detail previously.13 A double-blind pilot RCT was initiated in the community by research-trained paramedics from the North of Tyne division of the North East Ambulance Service (NEAS) NHS Trust. This division of NEAS covers a population of approximately 500 000 with 1200 suspected stroke admissions per year to three stroke units who participated in the trial. Of approximately 200 paramedics who work in North of Tyne, 76 volunteers attended a training day to take part in the trial. The training day included Good Clinical Practice training and Paramedic Initiated Lisinopril For Acute Stroke Treatment (PIL-FAST) specific procedures, including stroke recognition, consent and administration of study medication. Paramedics received payment and travel expenses for attending the training day.
The primary objective was to demonstrate whether it was possible to enrol at least four patients per month into the trial. This reflected the number of patients thought likely to fulfil the PIL-FAST eligibility criteria following an examination of stroke patient characteristics in the NEAS Database. The secondary objectives included determination of the proportions of suspected stroke patients potentially eligible for the study and those attended by a research-trained paramedic, compliance with data collection procedures, completeness of receipt of study medication and collection of clinical data (eg, BP) to inform the design of a definitive RCT.
Selection of participants and consent
Adults with new unilateral arm weakness thought to be due to acute stroke within 3 h of symptom onset and hypertension (systolic BP >160 mm Hg on two consecutive occasions) were eligible provided they were being transported to one of the three hospitals in the study and had no exclusion criteria that included contraindications to lisinopril and clinical features associated with stroke mimics. Eligible participants were identified, recruited, verbally consented and given the first dose of study medication by a research-trained paramedic attending the emergency call. A standardised consent script incorporating a simple capacity assessment was designed for use by paramedics (figure 1). Having listened to the study description, if the patient answered the study questions correctly, they were asked to provide verbal consent to participate in the trial. If the patient was unable to answer the questions or appeared to lack mental capacity during the paramedic routine assessment, the next of kin or other close relative (if present) could provide consent on the basis of their understanding of the wishes of the patient. Following arrival at hospital, patients and/or next of kin/close relative were given more information about the study and a standard information sheet. They were subsequently asked to confirm consent in writing. These unique consent processes were specifically designed to enable this trial to take place in the emergency setting without creating a delay in standard care.

Standardised paramedic consent script.
Further clinical data were collected following arrival at hospital and a review of appropriateness to continue on study medication was performed. This review allowed for data unavailable to paramedics in the community to be assessed prior to administration of further trial medication (eg, renal function). Participants discontinued from study medication continued in the trial for follow-up assessments.
Interventions
The protocol specified that participants receive 5–10 mg of lisinopril or matched placebo daily for 7 days. The first dose, 5 mg, was administered by a research-trained paramedic sublingually, due to the high frequency of swallowing problems in acute stroke. The 5 mg tablet was crushed in a syringe crusher with 3–5 mL of water prior to sublingual administration. Following arrival at hospital and review of appropriateness to continue study medication, dose titration was performed. If the systolic BP remained >150 mm Hg, a second 5 mg dose was to be administered and a daily dose of 10 mg was prescribed. If the systolic BP was <150 mm Hg, a second dose was not to be administered and a daily dose of 5 mg was prescribed. If clinically significant hypotension occurred, trial medication was withheld.
The PIL-FAST trial pack and randomisation
To enable paramedic access to study medication and administration equipment in the prehospital setting, boxes of medication (lisinopril or placebo, identical in appearance) and other study materials (paramedic research paperwork, syringe crusher, 5 mL syringe, 5 mL vial of water) were packaged into a ‘trial pack’. Each pack carried a unique study number according to the randomisation code (intervention and control in 1:1 ratio). One pack was issued to each research-trained paramedic, carried on emergency shifts and opened when an eligible patient consented to enter the trial. Opened packs accompanied the participant to hospital where the contents were collected by the hospital research team.
Data collection
Clinical data were collected at baseline, 3 and 7 days. Demography, medical history, BP, stroke severity (National Institute of Health Stroke Scale (NIHSS)14) and renal function were recorded. Timings of paramedic arrival on emergency scene, delivery of study medication and arrival at hospital were also recorded.
Data analysis
In accordance with recommendations for the analysis of pilot trials,11 ,12 a formal power calculation was not carried out, the data analysis is descriptive and statistical comparisons between the randomisation groups have not been undertaken.
Estimation of the proportion of patients potentially eligible for the trial
We used clinical ambulance and hospital records to report the proportions of stroke patients admitted to the trial hospitals who were eligible for the study, assessed by a PIL-FAST-trained paramedic and enrolled in the study, for the first 6 months of the trial. We were unable to review the complete trial period as a new paramedic record system was introduced during the study and problems searching these new records could not be overcome before the end of the study. Routine NEAS suspected stroke records were reviewed from 29 October 2010 to 30 April 2011. Eligibility was reviewed in a stepped approach such that if one eligibility criterion was not met, no further criteria were assessed for that individual.
Results
This pilot trial ran from 29 October 2010 to 15 December 2011. Fourteen participants were recruited (intervention group n=6; control group n=8). Ten participants (79%) provided their own verbal consent to the attending research-trained paramedic, for 1/14 (7%) consent was given by a next of kin/close relative and for 3/14 (21%) paramedics recorded that both the patient and a relative had given verbal consent. Baseline characteristics are shown in table 1. The median (IQR) admission NIHSS score was four (3–9), indicating that most participants had suffered minor—moderate strokes. Three participants (intervention group) received thrombolysis.
Baseline characteristics
Participant flow through the trial is shown in figure 2. Two participants withdrew from the trial. One participant withdrew following arrival at hospital and review of the patient information sheet, considering the side effects of lisinopril to be unacceptable. For the second participant, a relative provided initial verbal consent, but was distressed following arrival at hospital and unable to provide confirmation of consent in writing. Eleven (86%) participants completed study follow-up.

Paramedic Initiated Lisinopril For Acute Stroke Treatment flowchart.
Four of the 14 trial participants (28%) completed the full 7 days of study treatment. For 2/14 trial participants (14%), study medication was discontinued after day 3 because of a non-stroke diagnosis in one case and systolic BP <120 mm Hg in the second. The remaining 8/14 participants (57%) received the ambulance dose of study medication only for the following reasons: study withdrawal following arrival at hospital (n=2), loss of study medication in handover between paramedics and hospital staff (n=1), further medication withheld as systolic BP <120 mm Hg (n=1), discontinuation of medication by the hospital research team following the review of appropriateness to continue on arrival in hospital (n=4: incorrect judgement of stroke onset time by paramedics (n=3), physician decision not to continue with antihypertensive in a patient with intracerebral haemorrhage, even though this was not a reason to discontinue within the study protocol (n=1)).
Clinical outcomes are shown in table 2. One participant (control group) had a clinically important rise in serum creatinine (100 μmol/L) between baseline and day 7. The median time from paramedic arrival on scene to arrival at hospital for trial participants was 38 (IQR 32–42) min. This compares favourably with a median of 32 min for routine stroke admissions in the North of Tyne area in the year preceding the trial. The median time from paramedic arrival on scene to delivery of study medication was 25 (IQR 17–28) min. The median time from stroke onset (as assessed by paramedic) to delivery of study medication was 70 (IQR 40–89) min.
Clinical outcomes
Four Serious Adverse Events (SAEs) were reported (intervention group n=2, control group n=2). One was an episode of hypotension which occurred following the first dose of study medication (lisinopril) and was treated with intravenous fluids. Two were chest infections in patients with severe stroke (NIHSS 16 and 21) which resulted in death after the 7-day study period (intervention group n=1, control group n=1). There was one case of post-haemorrhagic obstructive hydrocephalus (control group).
There were few missing data for trial participants (tables 1 and 2). One paramedic recorded that she/he had dropped a study tablet on the floor and had used a second tablet to treat the participant, but no other issues with prehospital medication administration were reported.
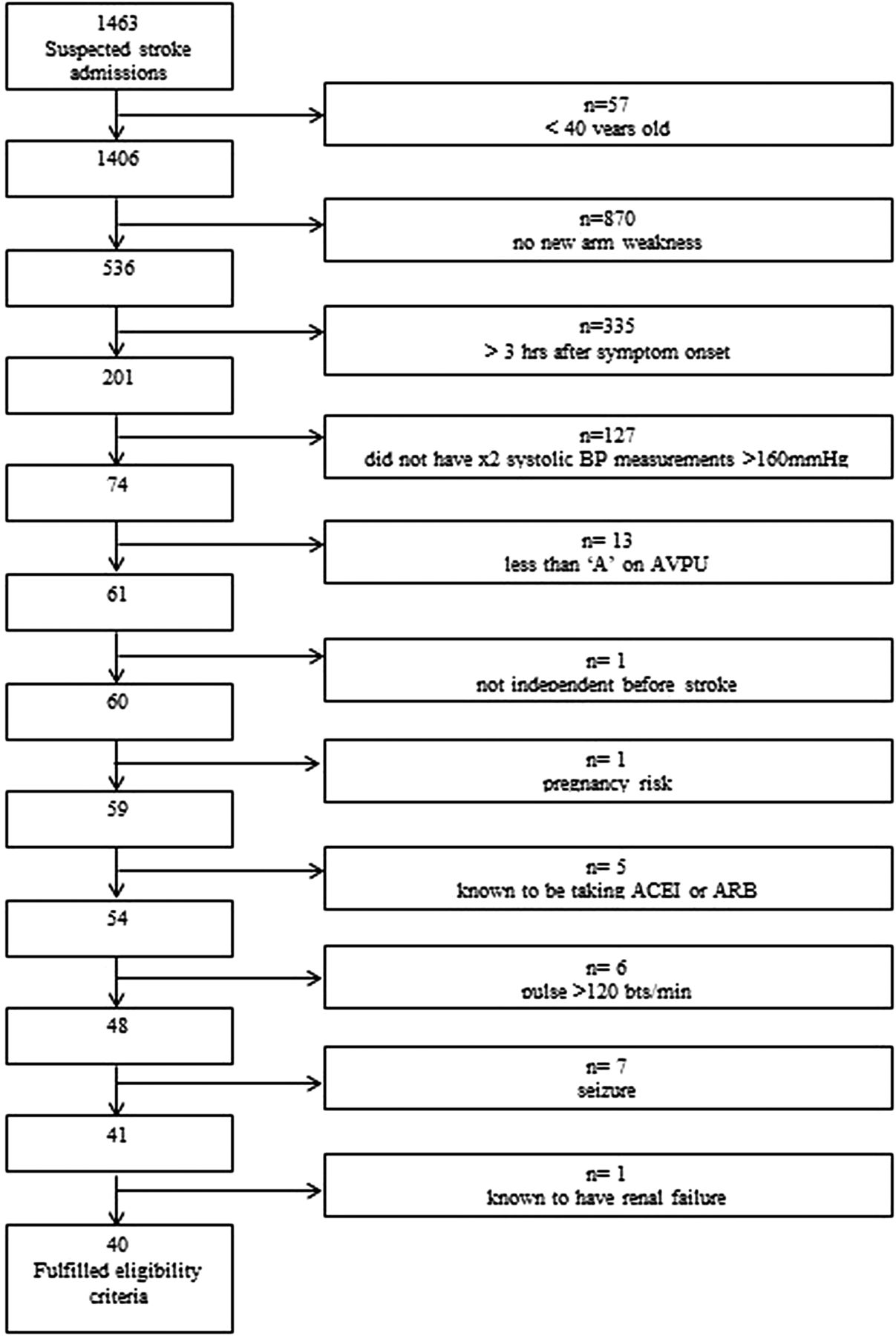
From 29 October 2010 to 30 April 2011, 1463 suspected stroke patients travelled by ambulance to the three trial hospitals. Forty of the 1463 (3%) suspected stroke patients fulfilled the PIL-FAST eligibility criteria and 13/40 (33%) were attended by a PIL-FAST-trained paramedic. In this 6 -month period, seven patients were enrolled in PIL-FAST. Therefore, 7/13 (54%) patients eligible for PIL-FAST who travelled with a PIL-FAST-trained paramedic were recruited. For the 6/13 patients who were eligible for PIL-FAST, travelled with a PIL-FAST-trained paramedic and not recruited, paramedic records indicated the following: considered for PIL-FAST but believed not to be eligible (n=1), considered for PIL-FAST but no further details given about non-enrolment (n=1) and no notes about PIL-FAST (n=4). The reasons for non-eligibility for the 1423/1463 (97%) suspected stroke patients are shown in figure 3.

{kind=link}
{kind=link}
{kind=link}
Reasons for non-eligibility for PIL-FAST. AVPU, Alert, Voice, Pain, Unresponsive; ARB, angiotensin receptor blocker.
Discussion
We have demonstrated that it is possible to conduct a paramedic initiated double-blind RCT of a treatment for acute stroke. Research-trained paramedics were able to identify eligible patients, obtain verbal consent, complete research paperwork and administer a sublingual dose of study medication. Research staff at the receiving hospitals were able to continue the trial protocol.
Recruitment was approximately one patient per month and therefore less than our target of four patients per month. However, the recruitment rate was similar in terms of recruitment/million population to that reported in the ongoing FAST-MAG trial15 and in hindsight, our recruitment target was too ambitious. Our review of routine paramedic records showed that approximately six to seven patients met the eligibility criteria per month (40 patients in 6 months), but only 33% of eligible patients (approximately two per month) were attended by a PIL-FAST-trained paramedic, of whom 54% were then recruited to the trial. Lack of recruitment of eligible patients may relate to paramedic confidence in stroke diagnosis and use of the PIL-FAST consent process, the ease of availability of information required for PIL-FAST on the emergency scene and general service pressures. However, the proportion of eligible patients recruited compares very favourably with inpatient stroke trials where our experience suggests at most 25% of eligible patients are recruited. To achieve a higher recruitment rate, it would be necessary to train more paramedics and to explore potential barriers to recruitment. All paramedics who took part in PIL-FAST were volunteers and it may not be feasible to expect greater numbers to take part in a future study unless research participation is seen as a core responsibility of the paramedic role. In preparation for PIL-FAST, focus groups of senior paramedics revealed enthusiasm for research into stroke treatments, but highlighted significant concerns about the impact on the length of the prehospital phase and professional boundaries.16 A recent survey of ambulance service staff in North America reported that 38% believe that they should retain the right to decide whether to support a particular study.17 If sufficient numbers of paramedics are to engage actively with a study, it will be necessary to offer reassurance that delays in access to hospital care can be minimised and to find mechanisms to encourage research support through learning credits, appraisal mechanisms and performance feedback.
Recruitment could also be improved by modifying the eligibility criteria. The review of paramedic records showed that a large number of suspected stroke admissions were ineligible for PIL-FAST due to the absence of unilateral arm weakness or symptom onset outside the 3 h. Extending the time window for inclusion and including patients with uncertain time of onset would result in more patients being eligible, but would defeat the purpose of an evaluation of very early treatment. Patients were required to have unilateral arm weakness as this is the most reliable symptom of acute stroke.18 Paramedics are not experts in the diagnosis of stroke and it was important that inclusion of stroke mimics was avoided where possible. While extending the symptoms of stroke which paramedics could use for inclusion is possible, this increases the chance of including non-stroke patients and patients with very mild impairments (eg, only facial weakness) who are unlikely to show any significant change in neurological status at outcome.
Enrolment into PIL-FAST did not result in a clinically important delay in transfer to hospital, and drug administration was a median of 70 min since paramedic assessment of symptom onset. In Los Angeles, the FAST-MAG trial has reported a median time from symptom onset to initiation of study medication of 46 min.19 The difference may be due to the more suburban and rural setting for PIL-FAST causing a longer arrival time for ambulances compared with urban Los Angeles.
The consent process designed for PIL-FAST appears to have been largely acceptable. As one patient withdrew consent after reading the patient information sheet in hospital, a future trial may require more information about side effects to be discussed prehospital. The second withdrawal was because proxy written consent for an incapacitated participant could not be obtained. As mental incapacity occurs more often with disabling stroke but these have potentially the most to gain from treatment, we were keen to include these participants. We did not think it acceptable to pursue a ‘research without consent’ approach as NEAS paramedics were naive to research and clinical trials but this approach could be considered for future trials involving agents with good safety profiles.
Only four of the 14 trial participants completed the full 7 days of study medication, which is an important feasibility issue. In 4/10 cases where treatment was not completed, this could be avoidable in a future study. In 3/4 of cases, the hospital stroke team considered that paramedics had made an incorrect assessment of stroke onset time and stroke symptoms had not started within 3 h which could be improved through training. However, this may also reflect the challenge of obtaining reliable information at the scene, and highlights the importance of having a review to continue with the study soon after arrival at hospital or having physician support by telephone to paramedics in the field. While paramedics could seek advice from on-call stroke specialists at any time of the day or night, this was not sought for any participants in our study. In 1/4 of cases, study medication was lost in the prehospital/hospital handover which should be avoidable but demonstrates a need to improve this process.
Limited conclusions can be drawn from the BP readings in this small pilot study, but our observations are consistent with the mean BP reduction of 14 mm Hg at 24 h after lisinopril administration compared with control seen in the Controlling Hypertension and Hypotension Immediately Post Stroke (CHHIPS) study.3 The Scandinavian Candesartan Acute Stroke Trial (SCAST)2 did not show any advantage to BP reduction within 30 h after stroke onset and there was a suggestion that it may be harmful to lower levels after the hyperacute phase, but patients were included with an initial systolic BP between 140 and 160 mm Hg, which would have excluded them from PIL-FAST. Although lacking statistical power, a subgroup analysis of SCAST suggested that patients treated between 0 and 6 h after symptom onset had a better outcome.2
The median baseline NIHSS score of participants was only four, which is lower than expected. This may mean that paramedics were unable or unwilling to recruit the more severely affected stroke patients. Due to the nature of stroke impairments, it would often not be possible to take consent from a severely affected patient and they would only be able to be included if a close relative was present and an onset time could be established.
In conclusion, we have shown that it is possible to conduct a paramedic initiated double-blind RCT of a treatment for acute stroke. However, to perform a definitive RCT in a reasonable timescale, a large number of trained paramedics across several ambulance services would be needed to recruit the number of patients likely to be required.
Acknowledgments
We thank the following for their contribution: patients who participated in the trial; local investigators (Ms Ann Fox, Dr Anand Dixit); PIL-FAST-trained paramedics; Stroke Research Network clinical trial officers who collected data in hospitals; pharmacy teams at Newcastle upon Tyne Hospitals NHS Foundation Trust and Lloydspharmacy; Newcastle Clinical Trials Unit (Gillian Watson and Chris Speed, trial management; Ruth Wood, data management); Trial Steering Committee (Professor Tom Robinson, Dr Gavin Young, Mr Paul Jackson, Mr and Mrs P Elliott) and Data Monitoring and Ethics Committee (Professor Simon Thomas, Dr Alex McConnachie, Dr Pippa Tyrrell).
References
Footnotes
-
Contributors CP and GAF conceived the study and obtained funding. LS, CP, SM, DH, EM and GAF further designed the study and wrote the study protocol. LS managed the trial. SM orchestrated the involvement of the North East Ambulance Service (NEAS). PY performed the retrospective review of potential study eligibility using NEAS paramedic records. DH was the study statistician. LS drafted the manuscript. All authors contributed substantially to revision of the manuscript. GAF takes responsibility for the paper as a whole.
-
Funding This article presents independent research funded by the National Institute for Health Research (NIHR) under its Programme Grants for Applied Research scheme (RP-PG-0606-1241). The views expressed in this publication are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
-
Competing interests GAF has received lecture fees and research funding from Pfizer, Boehringer-Ingelheim and Lundbeck A/S.
-
Ethics approval Newcastle and North Tyneside 2 research ethics committee.
-
Provenance and peer review Not commissioned; externally peer reviewed.