Article Text

Abstract
Objective To determine if intravenous paracetamol was superior to oral paracetamol as an adjunct to opioids in the management of moderate to severe pain in the ED setting.
Methods A prospective, randomised, double-blind, double-dummy, controlled trial was conducted at a single academic tertiary care ED. Adult patients with moderate to severe pain were randomly assigned to receive either the intravenous paracetamol or oral paracetamol. The primary outcome was Visual Analogue Scale (VAS) pain reduction at 30 min. A clinically significant change in pain was defined as 13 mm.
Results 87 participants were included in the final analysis, with a median age of 43.5 years and 59.8% were female. Overall mean baseline VAS pain score was 67.9 mm (±16.0). Both formulations achieved a clinically significant mean pain score reduction at 30 min, with no significant difference between the groups with 16.0 mm (SD 19.1 mm) in the intravenous group and 14.6 mm (SD 26.4) in the oral group; difference −1.4 mm (95% CI −11.6 to 8.8, P=0.79). Secondary outcomes, including postintervention intravenous opioid administration, patient satisfaction, side effects and length of stay, did not differ between groups.
Conclusions Overall, there was a small but clinically significant decrease in pain in each group. No superiority was demonstrated in this trial with intravenous paracetamol compared with oral paracetamol in terms of efficacy of analgesia and no difference in length of stay, patient satisfaction, need for rescue analgesia or side effects.
- pain management
- analgesia/pain control
Statistics from Altmetric.com
Key messages
What is already known on this subject
Intravenous paracetamol is increasingly used in EDs, despite limited and conflicting evidence of efficacy in this setting.
Despite a higher unit cost, intravenous paracetamol has potential advantages over the oral formulation.
What this study adds
This is the first comparison of oral with intravenous paracetamol as an adjunct to opioid analgesia in the ED setting.
We found a small but clinically significant reduction in pain with both intravenous and oral paracetamol, but there was no statistically significant difference found between the two formulations.
Introduction
Background
Pain is a common complaint of patients attending the ED, contributing to up to three-quarters of presentations and represents a significant burden on the healthcare system.1–4 Despite this, pain management in the ED has consistently been shown to be suboptimal5–7 and may contribute to poorer patient outcomes and patient dissatisfaction.
Pain management protocols involve initial pain assessment, provision of appropriate analgesia and regular pain reassessment.8 Opioids such as intravenous morphine are often recommended as first line for severe pain.8 Analgesics such as oral paracetamol, oxycodone and non-steroidal anti-inflammatory drugs are used as adjuncts or for less severe pain presentations. An intravenous preparation of paracetamol has been available for 10–15 years, although availability in EDs is often restricted.9 In the ED setting, intravenous paracetamol has been compared with intravenous morphine and other active comparators, with conflicting results.10–13 There is often an assumption of superior efficacy of the intravenous preparation due to the pharmacokinetic advantages including higher bioavailability, achieving target plasma concentrations faster and avoiding the hepatic first pass effect, but comparisons with the oral form are limited.9 14 Potential advantages of intravenous paracetamol over the oral formulation include faster onset of action, greater analgesic efficacy and reduced opioid use with less opioid associated side effects.14 15 Justification of the preferential use of intravenous paracetamol, when oral and per rectum routes of administration are available, with the prospect of higher costs, time and inconvenience and the possibility of complications with intravenous administration, requires evidence of superior clinical efficacy.
Objectives
The objective of the study was to compare the efficacy of intravenous paracetamol with oral paracetamol as an adjunct to intravenous opioids in an undifferentiated group of adult patients in moderate to severe acute pain in the ED setting.
Methods
Study design and setting
A single-centre, randomised, double-blind, double-dummy, active-controlled superiority trial was performed in the ED and short stay unit of a mixed (adult and paediatric) tertiary, academic ED in Australia with an annual census of over 85 000 presentations per year. The trial was registered with the Australian and New Zealand Clinical Trials Registry: Trial ID ACTRN12615000481572. Patient recruitment occurred from April 2015 to February 2016.
Selection of participants
Patients were eligible for inclusion if they were ≥18 years old, complaining of acute pain corresponding with a Visual Analogue Scale (VAS) ≥40 mm at 5 min after receiving at least one dose of intravenous opioid, as we felt it was unethical to delay opioid administration for patients with moderate to severe pain. Patients were excluded if they had any medical history that would impair accurate pain assessment at the clinician’s discretion, chronic pain, known or suspected opiate dependence, allergy to paracetamol, any degree of renal or hepatic insufficiency, pregnant, currently enrolled in other pain trials, unable to swallow oral medication or had taken paracetamol within the last 6 hours. Patient recruitment and enrolment was performed by research personnel and ED staff restricted to ED pharmacy hours (07:30 to 20:00 Monday to Friday and 07:30 to 17:30 on weekends).
Interventions, randomisation and masking
The trial used a double-blind, double-dummy technique in that all patients received both an oral and an intravenous preparation. Patients were allocated to active 1 g (2×500 mg) paracetamol (Panadol, GlaxoSmithKline, Ermington, New South Wales, Australia) or identical placebo orally AND active paracetamol, 1 g intravenous (1 g per 20 mL) (Actavis, Macqurie Park, New South Wales, Australia) or normal saline, with both intravenous formulations administered in 100 mL normal saline over 15 min. The oral intervention was manufactured by an external good manufacturing practice (GMP) licensed facility specifically for this study so that placebo and active paracetamol were identical in appearance, taste and smell. The intravenous intervention was drawn up by the unblinded pharmacy study staff into visually identical 100 mL normal saline bags and generically labelled as ‘Paracetamol trial - IV paracetamol/placebo’.
The randomisation sequence was produced using a computer-generated program of permuted blocks with varying block size, in a simple 1:1 allocation ratio with no stratification by a clinical trial pharmacist. A sealed envelope technique was used to maintain allocation concealment. Once eligibility was confirmed by clinical or research staff, pharmacy was contacted by telephone and would select the next corresponding envelope for the allocated intervention. Pharmacy staff would then proceed to prepare the intravenous and oral interventions in a separate private room with a closed door, to where the patient was being treated. Both intravenous and oral preparations were then provided to the treating nurse for administration. The pharmacist had no involvement in patient care or data collection. All participants, study staff and treating clinicians were masked to treatment allocation for the duration of the study and data analysis.
Patients provided verbal consent or assent prior to participation, and formal written consent was deferred until pain was adequately controlled and patients deemed as having capacity to provide valid informed consent. Patients discharged from ED prior to obtaining written consent were mailed a consent form to return to investigators. This process was approved by the hospital Human Research and Ethics Committee. Screening logs of potentially eligible patients were not kept.
Methods of measurement
Baseline data including VAS score, HR, RR, BP, oxygen saturation and GCS were entered onto patient case report forms at enrolment. Demographic details were recorded from hospital databases including gender, date of birth, triage time, chief complaint and cause of pain characterised as: abdominal/gastrointestinal, musculoskeletal, neurological and other.
Pain scores were assessed using the VAS, a validated tool for ED pain research.1 Participants recorded a mark on the 100 mm VAS line to represent a pain score, labelled with ‘0=no pain’ and ‘100=worst possible pain’. Vital signs, adverse event data and VAS scores were collected on a specifically designed data collection instrument immediately prior to the administration of the study drugs (t=0), then at 15 minutely intervals for the first hour and half hourly intervals for the subsequent 3 hours or until the patient was transferred to the ward or discharged. Other medications, doses, routes and timing of administration were recorded. Patient satisfaction with overall pain management was ascertained as a percentage (0%–100%) with higher scores representing higher satisfaction, at the conclusion of data collection. Data were collected by clinical staff or dedicated research staff when available.
All management after the administration of the study medication was at the discretion of the treating clinician, including the administration of further doses of opioid analgesia according to hospital protocols.
Outcomes
The primary outcome was the reduction of pain score on the VAS from baseline at 30 min, as a clinically meaningful endpoint in which to assess pain for an ED study.16 Secondary outcomes were mean VAS score reduction at other time points, occurrence of adverse events, rescue analgesia administered (morphine equivalents in milligrams), patient satisfaction and length of ED stay. An additional secondary outcome of the proportion of patients achieving a clinically significant reduction in pain score in each group (defined as a reduction of VAS pain score of >50% of baseline) was included.
Statistical analysis
Power calculation determined that 44 patients were required in each group with 80% power and a significance of 0.05% to detect a clinically significant difference in mean pain scores of 15 mm, assuming an SD of 25 mm. The difference of 15 mm represented a compromise between the commonly cited ‘minimum clinically significant’ difference of 13 mm17 and 20 mm.6 ,18 To allow for attrition and withdrawal, recruitment was aimed at a total of 100 patients to be split evenly between trial groups.
Data were analysed using SPSS V.22.0 (IBM, released 2013, IBM SPSS Statistics for Windows). The primary analysis was performed on a modified intention-to-treat principle, including only patients meeting the prespecified moderate pain criterion of >40 mm (figure 1) at time point t=0. Data on all patients are also presented. Descriptive statistics were used to describe the baseline characteristics of the study population. A one-way analysis of covariance was conducted to compare the effectiveness treatment on VAS scores. Levenes test and normality checks were carried out and the assumptions met. Differences between groups determined using Bonferroni test adjusted for multiple comparisons. Repeated measures analysis of variance (ANOVA) was used to compare pain scores at different time points. Categorical data were analysed using the χ2 test. Some authors have questioned whether smaller reductions of pain score are ‘clinically meaningful’, particularly for more severe pain at the upper end of the VAS.19 Therefore, we performed an additional analysis of the proportion of participants who achieved a clinically meaningful reduction in pain at 30 min, in both groups, defined as a 50% reduction from baseline. Proportions were compared with χ2 test. For all analyses, a P value <0.05 was considered statistically significant.

Consolidated Standards of Reporting Trials participant flow diagram. mITT, modified intention to treat; VAS, Visual Analogue Scale.
Results
Characteristics of study subjects
A total of 142 patients were enrolled in the study, 34 who did not have documented written informed consent prior to being discharged from the department and did not return the mailed written consent form, thus were excluded from the analysis, leaving 108 with valid written consent and available for analysis. Of these patients, 87 had a pain score >40 mm at t=0 and were included in the primary modified intention-to-treat (mITT) analysis20: 47 in the intravenous group and 40 in the oral group (see Consolidated Standards of Reporting Trials flow diagram, figure 1).
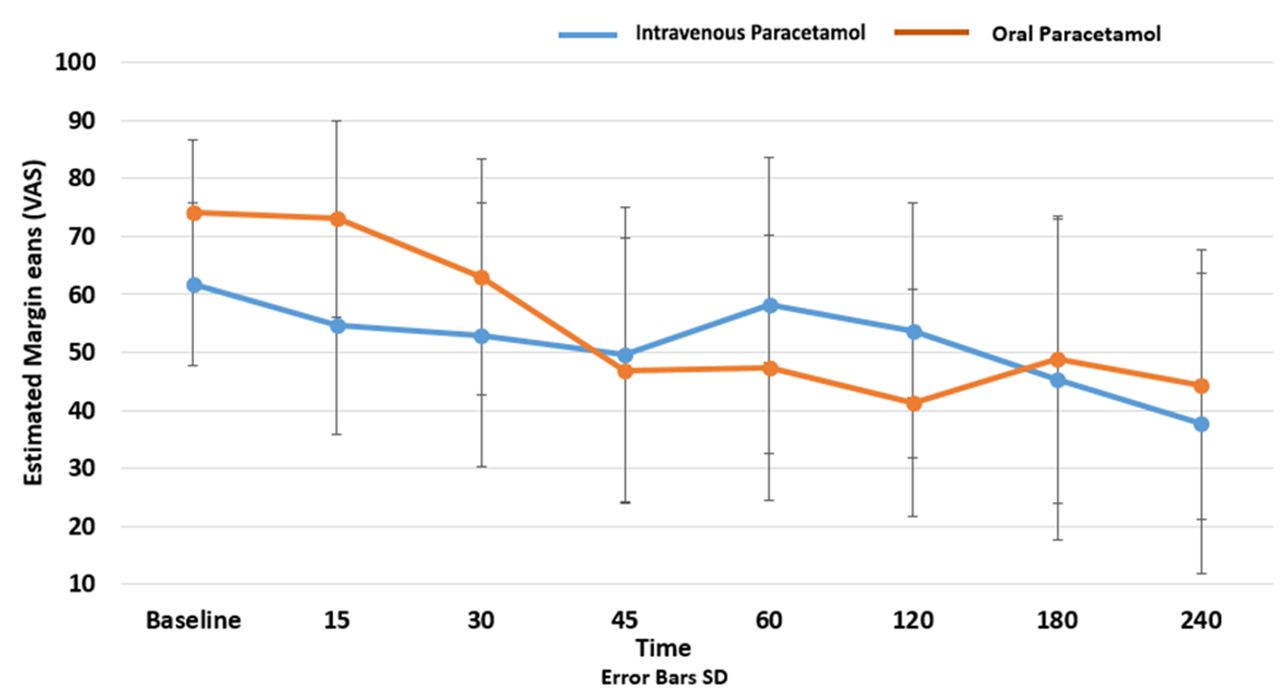
Baseline characteristics were similar in each group (table 1), and mean pain scores at baseline (t=0) were 65.0 (15.8) in intravenous group and 71.3 (15.6) in oral group. Mean pain scores and SD at 30 min were 51.5 mm (22.9) in the intravenous paracetamol group and 54.2 mm (23.1) in the oral group, respectively (table 2). For the primary outcome of pain score reduction at 30 min, difference in pain score reduction was −2.6 mm (95% CI −13.2 to 7.9, P=0.62). Repeated measures ANOVA was conducted to investigate the impact of treatment with time on pain scores. There was no significant main effect for time, Wilk’s Lambda 0.28, F (7,91), P=0.12. There was no significant interaction effect between time and treatment groups, Wilk’s Lambda=0.43, F(7,91), P=0.36. There was no significant difference between the groups reduction of pain scores, F (1,13)=0.08, P=0.77, partial eta 0.006 (figure 2).
Baseline characteristics (modified intention to treat)
Pain scores at various time points (modified intention to treat)
{kind=link}
{kind=link}
Change in VAS scores over time. VAS, VAS, Visual Analogue Scale.
For secondary outcomes, ‘rescue’ opioids were administered in 106 instances in 86.2% of the intravenous group compared with 122 instances in 84.1% of the oral group (P=0.69), with a median and IQR dose of 5 mg (2.5–5) morphine equivalents in the intravenous group and the oral group 5 mg (2.5–5.0) equivalent of intravenous morphine (P=0.50). Patient satisfaction in the intravenous group was 100.0% (80.0–100) and 90.0% (75.0–98.0) in the oral group (P=0.10). Median (IQR) length of stay in minutes was also similar between groups: intravenous 195 (153–232) and the oral group 203 (63–227), difference −8 min (P=0.95). Side effects were infrequent with only one report of tachycardia at 240 min postintervention in the oral group and one brief episode of hypotension reported at 45 min in the intravenous group, both not thought to be due to the intervention. Time to administration (triage time to intervention administration time) was similar in both groups, median in minutes (IQR): intravenous 80 (59–108) and oral 74 (58–100) (P=0.35) (table 3).
Secondary outcomes
In the additional analysis, we found that 25.5% (n=12) in the intravenous group and 20.0% (n=8) in the oral group achieved significant pain reduction as defined, difference of minimum 50% at 30 min (P=0.54) χ2 test. There was no difference at time points 60 and 90 min (online supplementary appendix). Results for all consented randomised patients (n=108) did not differ substantially from those of the mITT group (online supplementary appendix).
Supplementary file 1
Limitations
Our study had a number of important limitations. First, our study enrolled a convenience sample of ED patients with acute pain, which may have resulted in selection bias. ED pharmacy staff prepared study medications, as the planned manufactured placebo intravenous preparation was not possible due to difficulty in obtaining a stable and sterile product identical to the active preparation and varying escalations in pricing between the time of securing funding and commencement of the study. Consequently, a pure identical intravenous placebo preparation was deemed not feasible, and the study team decided to unblind the pharmacy members to progress the trial. This also resulted in some delay in the administration of the intervention, which may have influenced the effectiveness of the intervention. Measurements were taken from the time of administration (t=0) rather than when ordered, which might have negated time advantages of more rapid oral administration. The intravenous formulation was administered over 15 min, compared with immediate administration of the oral formulation. Although this seems to favour the oral group, this is reflective of usual clinical practice.
Patients were excluded from analysis if they did not have a valid written informed consent. Patients provided verbal, implied consent and assent to study participation during the course of the trial; however, the ethics committee prohibited inclusion of data without written informed consent. This was comparable in both intervention groups, and baseline characteristics were similar to included patients. While this may have resulted in a loss of power, we do not believe this would have introduced systematic bias favouring either group. There were some minor imbalances between groups at baseline, which might have introduced some confounding of results. Some ‘eligible’ patients had improvements in their pain scores prior to the delivery of the intervention. We only included patients who had received opiate analgesia, as we did not feel it ethical to withhold analgesia from a group for the purposes of a study including a placebo arm. Our study used the VAS as a highly reliable and valid pain assessment tool in ED research.21 22 However, pain is a very subjective experience and it is possible the VAS did not capture all clinically important data. Due to resource limitations, we assessed patient satisfaction crudely on a scale of 0%–100%, which is not a validated tool, and we acknowledge that other aspects of care, such as regular pain assessment, may have influenced satisfaction in both groups. Finally, the study was conducted at a single centre, and so there is a limit to the generalisability of the results of this study.
Discussion
This is the first study comparing intravenous and oral paracetamol for the reduction of pain in the ED setting. Our results found both preparations to have only modest analgesic efficacy, and we found no superiority of pain reduction for the intravenous preparation in patients with moderate to severe pain.
Some previous research has indicated that intravenous paracetamol has a faster onset of action, greater analgesic efficacy and longer duration of action, and these conclusions informed our hypothesis based on the biological plausibility.23 24 Although the intravenous preparation is often anecdotally regarded as superior, our results demonstrating no difference are congruent with a recent systematic review comparing oral and intravenous paracetamol in a variety of settings.9 These authors included six randomised trials in the review, only three of which assessed efficacy outcomes and all were in the postoperative setting. They found no clinically significant difference in efficacy or other outcomes. However, these conclusions were based on low-quality evidence overall, with methodological limitations in all included trials, such as issues with blinding and allocation concealment.
Intravenous paracetamol has also compared favourably with intravenous morphine in blinded, randomised controlled trials in ED settings for renal colic, low back pain and traumatic limb pain, although none of these studies could claim superiority of intravenous paracetamol.10–12 25 A recent systematic review found ‘limited evidence’ to support the use as a primary analgesic in ED setting when compared with opioid and other active comparators.13 In our study, we used oral paracetamol as the comparator, as we were assessing the efficacy of paracetamol in these two formulations as an adjunct to opioid analgesia regimes rather than a replacement.
There are a number of possible reasons for the intravenous preparation not demonstrating a superior effect over its oral comparator. The first is that no clinically meaningful difference in efficacy exists or be the result of a type II error. Alternatively, heterogeneity in our patient population or prior administration of opioid analgesic may have obscured any real treatment effect. Later time points in our study suffered from lack of power, with fewer patients remaining in the analysis. The ‘National Emergency Access Targets’ in use at the time required that 90% of patients leave the ED for admission, discharge or transfer within 4 hours,26 thus constraining access to longer-term data.
Historically, the primary barrier to use intravenous paracetamol has been one of cost. Recently, however, the patent for intravenous paracetamol has expired and it is now being produced generically, making it more affordable and accessible. Previously, intravenous paracetamol was priced at $A12.22 per 1 g dose and is now available for $A1.76 per 1 g dose (as per I-pharmacy at The Townsville Hospital at time of writing). Considering that the average cost of an ED presentation is between $A1037 if admitted and $A422 if discharged from ED,27 in this context, the cost of one dose of intravenous paracetamolis insignificant in real terms and becomes an option for this cohort of patients if beneficial. However, intravenous administration is associated with other potential disadvantages including increased time and inconvenience, indirect costs and potential side effects like local infection and phlebitis. The absence of benefit makes routine preferential use of intravenous paracetamol difficult to justify.
It is important to stress that our study design excluded patients who were not able to take oral medications. There may be patient groups in whom oral medication is not appropriate or contraindicated where intravenous paracetamol may still be an effective analgesic adjunct. Further research may explore patients with more severe pain at baseline or specific subgroups of ED patients such as traumatic pain.
In summary, our study did not demonstrate the superiority of intravenous paracetamol compared with oral paracetamol as an adjunct to opioids in a heterogeneous group of adult patients with moderate to severe pain in the ED. While these results do not support the routine use of intravenous paracetamol in this patient group, there may be patients that fall outside the boundaries of our eligibility criteria whom may benefit.
Acknowledgments
The trial was supported by a project grant from the Emergency Medicine Foundation. JF is supported by a scholarship from the Emergency Medicine Foundation. The authors would like to acknowledge the contribution of the Townsville Hospital and Health Service, the emergency department for supporting the trial and research assistants; Kirsty Freeman, Bethany Roche, Susan Montgomerry and Leonie Jones for their assistance with the trial. Also, a sincere thank you to all participants for sharing their time and experiences.
References
Footnotes
Contributors JF, DL and RR conceived the study and designed the protocol and funding application and conduct of the trial. BC assisted with funding application. VNV assisted with statistical design and analysis. KL, MF and KR assisted with design and study conduct including patient recruitment. JF was responsible for drafting the manuscript; all authors contributed to revisions and approve the final manuscript. JF takes responsibility for the work.
Funding Supported by grants from James Cook University and the Emergency Medicine Foundation. Grant number: EMPJ-107R22-2014EMPJ-107R22-2014.
Competing interests None declared.
Patient consent Obtained.
Ethics approval The Townsville Hospital and Health Service Human Research Ethics Committee (HREC/14/QTHS/151).
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Study data will be made available from the corresponding author on request.
Presented at Presented at the Australasian College for Emergency Medicine, Annual Scientific Meeting, Queenstown, New Zealand, November 2016.