Article Text

Abstract
Objective The current study aimed to ascertain differences in early postmedication pain reduction in participants presenting with acute musculoskeletal injuries (MSI) to the ED receiving intramuscular (IM) versus per oral (PO) diclofenac.
Methods This was a prospective, double-blinded, randomised controlled trial conducted between January and June 2018 at the ED of Hamad General Hospital in Doha, Qatar. Adults (18–65 years of age) presenting to the ED within 24 hours of an acute MSI, who had a triage pain score measured using numerical rating scale of at least five or above were enrolled in this trial. Participants randomised to either IM (75 mg) with oral placebo, or oral (100 mg) diclofenac group with IM placebo using a computer-generated randomised concealed list in blocks of six and eight. The primary objective was to compare the proportion of IM versus PO participants attaining a 50% reduction in pain score at 30 min from t0.
Results 300 participants were enrolled (150 in the IM diclofenac group and 150 in the PO diclofenac group) in the trial. The primary outcome was achieved in 99.3 (95% CI 96.3 to 100) in the IM group and 86.7 (95% CI 80.2 to 91.7) in PO group. There was an absolute risk difference of 12.7%. This corresponds to a number needed to treat of 8 cases (95% CI 6 to 14) receiving IM rather than the PO diclofenac in order to achieve one additional case of 50% pain reduction within 30 min of drug administration. There were no adverse events experienced in any treatment groups.
Conclusion IM diclofenac injection provides rapid analgesia over PO administration of diclofenac. However, given the preparation needed for an IM injection, oral administration may be preferable when and if clinical circumstances allow a choice in non-steroidal anti-inflammatory drug administration route.
- pain management
- emergency department
Statistics from Altmetric.com
Key messages
What is already known on this subject
Previous studies comparing different routes of administration for non-steroidal anti-inflammatory drugs (NSAIDs) suggest that the oral route is non-inferior to the intramuscular (IM) route for achieving effective analgesia. However, these studies have used two different types of NSAIDs.
There is a lack of evidence regarding comparison of a single NSAID compared via oral and IM route in a methodologically-sound, large clinical randomised controlled trial (RCT).
What this study adds
This RCT compared the early postmedication effect of diclofenac administered through IM versus oral route.
The IM administration of diclofenac was associated with a higher success for achieving 50% reduction in initial pain and overall efficacy in the initial 30-min postmedication (absolute risk difference 12.7%).
However, the absolute difference between the two groups for primary outcome comparison suggests that the per oral route is clinically non-inferior to IM route.
Introduction
Background
Pain alleviation is a well-established clinical priority in emergency medicine (EM).1 Recent years have seen ED practice shift towards increased use of non-opioids such as non-steroidal anti-inflammatory drugs (NSAIDs) and paracetamol, which do not have opioids’ dependency-related risks.1–3
Diclofenac is an NSAID commonly used in the ED for conditions such as musculoskeletal injury (MSI) and renal colic.4 5 Administered in adult doses ranging from 50 to 100 mg, diclofenac mediates analgesia primarily via inhibition of cyclo-oxygenase-2 (COX-2) enzymatic conversion of arachidonic acid into inflammatory prostaglandins. Diclofenac may be administered via more than one route. In the ED, the most common forms are per oral (PO) and intramuscular (IM).
Varying efficacies of different analgesia administration routes have previously been assessed in the ED. Studies comparing intravenous (IV) versus oral (PO) paracetamol administration did not find clinical superiority of IV route over PO.6 7 Classical evidence suggests little difference in pain relief for PO versus IM NSAIDs.8–11 However, similar work comparing single NSAID drug via two different routes of administration is lacking in the literature.
While existing data suggest IM and PO NSAIDs ultimately reach the same pain reduction endpoint, studies to date tend to include just one or two pain assessments in the early (first half-hour) postadministration time frame.8 9 11 With the EM evidence base suggesting that ultimate pain relief is equivalent between PO and IM NSAIDs, there is a remaining question as to the degree—if any—to which IM administration’s early effects are more pronounced. Even in the early postadministration time frame, better IM performance would be important to elucidate; recommendations from professional societies (eg, UK’s Royal College of EM12) acknowledge the evidence of clinical relevance to faster pain reduction even within the first half-hour of drug administration.13–18
Objectives
The objective of the study was to compare the efficacy and safety of IM diclofenac with oral diclofenac in achieving 50% reduction in pain at 30 min in patients with acute MSI in the ED.
Methodology
Study design and setting
This was a prospective, double-blind, double-dummy, randomised controlled trial (RCT) conducted at the ED of Hamad General Hospital in Doha, Qatar; the ED has with an annual census of approximately 450 000 patients.
Selection of participants
Eligible patients were healthy adult’s aged 18–65 years presenting to the ED with a soft tissue injury that occurred in the prior 48 hours, and had a pain score of 5 or more on a numerical rating scale (NRS). Patients were excluded who had taken NSAID or any pain killers within the past 12 hours, were allergic to diclofenac, had bronchial asthma, renal impairment, bleeding diathesis, known fractures; were pregnant, or when the clinical judgement of the treating physician was that the patient should not participate (eg, based on need to be nil PO).
Interventions, randomisation and masking
The participants were consecutively selected for enrolment in order of appearance during clinical shifts when investigators were available over the first 6 months of 2018. The subjects enrolled in the study were randomly assigned to either one of the study trial packets in a (1:1) ratio to receive either of the two combinations. Combination 1 was (active) 75 mg IM diclofenac injection with oral placebo tablets; and Combination 2 was (active) 100 mg oral diclofenac tablets with placebo IM injection. The study’s placebos were identical in appearance to the active drug. The computer-randomised sequence was generated within the block of six and eight and patients were allocated to one of the abovementioned combinations by the project coordinator who had no further role in the study. The subjects, clinicians and the data collector remained unaware of the treatment allocation. The statistician was also masked to treatment allocation. Allocation sequence was maintained within a sealed envelope. Once the subject was enrolled, the research associate obtained a written consent from the participant. Subjects were informed that they could drop out of the study any time, and receive rescue medication as needed, as well that the NSAIDs may take up to a half-hour to have effect and rescue medication was allowed by the study protocol after the 30-min NRS assessment.
Methods of measurement
The subject’s baseline vital signs and the demographic details were recorded. Subjects rated their pain using the NRS, a validated tool for pain assessment.19 It is an 11-point scale starting from (0 to 10) with 0 referring to no pain and 10 to be the worst pain ever. The initial pain score was recorded at t0 immediately before the administration of the study drug. Once the study drug was administered, the pain scores were recorded every 5 min for 30 min (t5, t10, t15, t20, t25 and t30) and then every 15 min until 60 min (t45, t60) had elapsed. The study’s monitoring also included assessment for side effects. Once complete data were obtained until 60 min, patient was handed over to the emergency physician on duty, for further care and disposition.
Outcomes
The primary outcome was defined as a 50% reduction of pain at 30 min from the baseline t0. The secondary outcome was defined as a minimum 2-point NRS drop from t0 till t30. Additionally, subjects were monitored for 1 hour after drug administration to assess for any adverse events.
Sample size
The sample size was calculated using 95% power, 5% level of significance, to detect pain difference, based on prior literature where if the primary outcome observed in the intervention group was 70%5 and in the control group was 50%.20 A total of 300 participants were required to detect a 20% difference between the groups for treatment success defined as 50% or more reduction in pain.
Statistical analysis
Information on study patients was collected initially using standard spreadsheet; data were transferred to the statistical analysis package Stata V.15MP. Stata was used for all analysis and graphics generation, with waterfall plots depicting pain reduction from baseline generated using coding as described by Schriger.21 Significance was set at the p<0.05 level.
The unit of analysis was the individual study subject. No patient was enrolled more than once. For NRS data, which were non-normal by Shapiro-Wilk testing, central tendency was reported as median with binomial exact 95% CIs calculated using Stata’s conservative method (ie, forcing limits to fall on sample values). The p values were calculated using the Kruskal-Wallis equality of population rank test.
All analyses were done as intention to treat. For categorical data, which included the study’s primary and secondary endpoints, results were calculated as proportions with binomial exact CIs at the 95% level. When point estimates were either 0% or 100%, CIs were reported at the one-sided 97.5% level. Fisher’s exact testing was used to evaluate statistical significance.
Characteristics of study subjects
A total of 300 subjects, 150 in each group (IM and PO), were enrolled from January 2018 through June 2018 (figure 1) of whom 78.3% were male. Patient characteristics per group are shown in table 1.
Patient characteristics
Pain score changes over time
All cases received the agent to which they were allocated, and no rescue analgesia was provided until after the 30 min from NRS assessment. All subjects had their pain assessed at each of the study’s nine NRS time frames. No subject dropped out of the study.
The changes in pain scores from baseline to each of the nine assessment points are reflected in figure 2. In each of figure 2’s plots, one vertical arrow represents a single-study subject. The figure’s mirrored arrangement of IM and PO subjects’ baseline NRS facilitates visualisation for symmetry in NRS changes for the two study groups.

Trial profile.
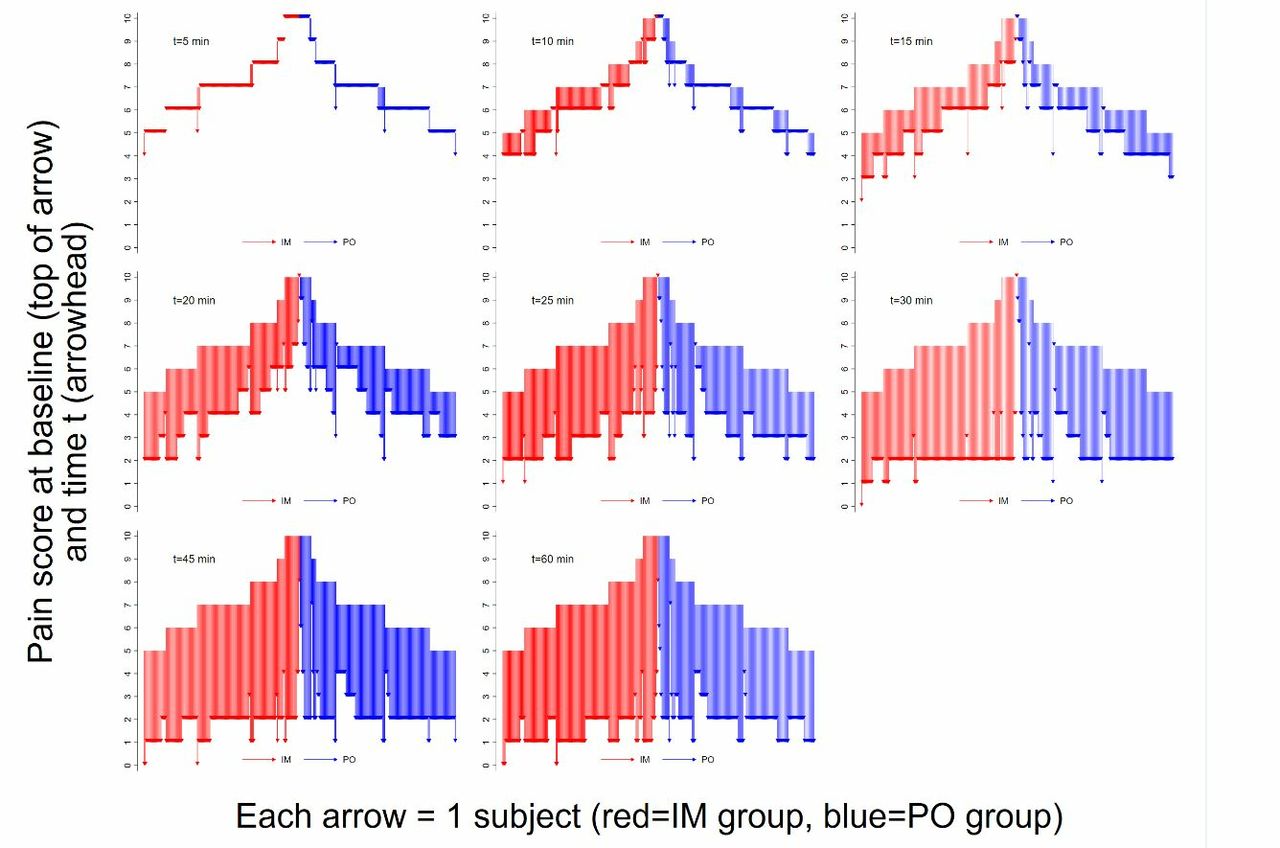
Waterfall plot of pain score decreases at each of the study’s timed assessments. IM,intramuscular; PO, per oral.
Table 2 reports the subjects’ pain scores as well as the proportions of IM and PO groups reaching the primary and secondary endpoints at each assessment time. Table 2 depicts an absolute risk difference of 12.7% for the primary endpoint (ie, 99.3% vs86.7%). This corresponds to a number needed to treat (NNT) of 8 cases (95% CI 6 to 14) receiving IM rather than the PO diclofenac, in order to achieve one additional case of 50% pain reduction within a half-hour of drug administration.
Pain score reductions and study endpoints
The proportions of IM and PO subjects reaching the primary and secondary endpoints are demonstrated in figure 3 and figure 4. Each figure shows the percent (with 95% CI) of subjects reaching the endpoint at a given assessment time.

Proportions of subjects (with 95% CIs) in each study group with pain reduction at least 50%. IM, intramuscular; NRS, numeric rating score; PO, per oral.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Proportions of subjects (with 95% CIs) in each study group with minimum 2-point drop in numeric rating score (NRS). IM, intramuscular; PO, per oral.
No patient experienced adverse events (one-sided 97.5% CI 0% to 2.4% for each group of n=150).
There was no rescue medication administered prior to evaluation of the primary and secondary endpoints at t30. In five cases, rescue medication was administered after assessment at t30; one case was in the IM group and four were in the PO group (p=0.371). The NRS data were reanalysed after the exclusion of the five cases receiving rescue medication; there were no changes in either the median or its 95% CI for the two relevant time points (t45 and t60) in either the IM or PO groups.
Discussion
This study was conducted to evaluate the degree to which administering diclofenac IM, rather than PO, increases likelihood of early (within a half-hour) achievement of 50% pain reduction. The results indicate that the IM route does offer some advantages over PO administration. However, this study found a relatively small margin of improvement associated with IM rather than PO diclofenac administration in the ED population examined. As was shown in table 2, central tendencies for NRS are essentially the same even when checked at frequent intervals in the early postadministration time frame. The timing advantage of diclofenac’s IM administration route is about 5 min, as demonstrated by figure 2’s waterfall plots and the small degree of ‘left shift’ of the IM versus PO diclofenac efficacy curves in figures 3 and 4.
In terms of overall efficacy, the primary endpoint was reached more commonly—nearly 100% of the time—with IM diclofenac, but the PO route also performed quite well (reaching the endpoint in 87% of cases). The NNT calculation means that in seven out of eight cases, PO diclofenac can be expected to reach the same endpoint of early 50% pain reduction as would be achieved with IM diclofenac.
This study’s main finding, that PO diclofenac works nearly as quickly and effectively as IM diclofenac, is perhaps counterintuitive since patients and physicians may presume that an injection works faster than a pill. Although the presumption of more rapid analgesia with IM NSAID use runs counter to some prior literature, the older evidence base could fairly be judged to include suboptimal methodology (eg, few assessments of pain in the initial half-hour after drug administration).8 9 11 Investigators have judged that pain relief rates within the first half-hour of drug administration are clinically important,22 and there are data suggesting that IM diclofenac decreases pain as quickly as 5 min after injection.23 In an era of increased emphasis on rapid alleviation of pain in the ED, clinicians wishing to provide the best care may not wish to wait a half-hour for PO analgesia to take effect and thus order the IM route.
However, the slim time advantage seen with the IM route comes with disadvantages associated with injection use. Some of these disadvantages are associated with time costs that can offset or even reverse the faster analgesia time seen with IM.5 In practical terms, it is likely that in the actual ED care setting, a PO analgesic can be administered at least 5 min faster than an IM analgesic, thus offsetting the IM route’s time gains seen in this study. The PO analgesic can be given with minimal drug preparation and nursing time, and does not require patient privacy to expose an injection site. Furthermore, the PO analgesic does not incur risks of injection-site complications that are either common (eg, pain) or rare (eg, tissue damage). Particularly in settings in which obtaining sterile needles is a non-trivial consideration, PO analgesia administration route can conserve resources. Even in locales where supplies are not an issue, the costs associated with IM NSAID administration are higher than those associated with administering a PO NSAID.
Of course, PO analgesia administration is not always practical or even possible. EM clinician choice of administration route should be informed by multiple factors. Clinical circumstances such as vomiting may dictate the need for parenteral analgesia. However, there is often a legitimate choice between IM and PO NSAID routes in the ED. It is likely that analgesia injections are sometimes employed due to perception that this route results in earlier, if not more profound, pain reduction. Such perception seems rational, but the existing EM evidence base and the current data argue against routine use of IM NSAIDs when PO NSAIDs are a practical alternative.
The current study is characterised by limitations that must be considered when interpreting the findings. First and most importantly, is the study’s setting in a site with a largely young male population from varied countries in the region. The study patients’ demographics may have contributed to the near-universal achievement of the prespecified ‘significant analgesia’ endpoint that is not usually seen in NSAID ED analgesia studies.22 24 However, the high rates of effective analgesia seen in the current analysis were similar to those from another (renal colic) analgesia trial performed at the study institution,5 and high analgesia rates have also been seen in other studies from southwestern Asia.25 The high efficacy of NSAID analgesia found in the current study should be considered applicable only to study populations that mirror that of the trial site and demographics.
Other study shortcomings are related to methodological approaches. The primary endpoint of 50% pain relief was selected as a reliable marker of substantial improvement in pain. This endpoint, previously utilised in an analgesia investigation at the study centre,5 is defensible as a representation of substantial pain reduction, but it is acknowledged that other endpoints (eg, complete pain relief) could have also been selected. The study’s secondary endpoint, achievement of a two-point NRS change, is consistent with the general tenor of pain reduction seen in many ED studies,26 but the utility of this endpoint was reduced by the fact that it was reached nearly universally in both IM and PO groups.
Despite the practicality of focusing on the most clinically relevant (early) time frame for ED analgesia, the study’s restriction of endpoint assessment to 30 min is also a limitation. Whereas diclofenac is expected to have a single-peak absorption when administered parenterally, PO administration of the drug may show dual-peak concentrations due to pH-dependent dissolution in the upper gastrointestinal tract.27 Continuation of pain assessments after 60 min, through the initial few hours would have provided a more complete picture of pain reduction (and possibly relapse) associated with more evidence suggesting IM route more effective than PO diclofenac.
As depicted in table 1, the two study groups appeared to be similar. There seems relatively low chance of confounding of the association between diclofenac administration route and the pain reduction endpoints. However, there is a risk of confounding due to unmeasured variables that affect diclofenac’s PO absorption (eg, fasting state).28
Although the study’s incorporation of different diclofenac dosages for the IM and PO routes mirrored common clinical practice, there is a risk that the lower IM dose (75 mg vs 100 mg for the PO route) biased NRS findings against the parenteral drug form. However, the use of the commonly applied ceiling doses for each administration route brings real-world results applicability that offsets the risk of bias in using different doses for IM and PO administration.
Finally, the long-term complications related to diclofenac such as renal failure, gastrointestinal bleeding and IM abscess were not recorded in this trial. In the current trial, we looked at immediate complications only; however, earlier trial conducted at the study centre including 1645 renal colic patients did not find any serious complications for using IM diclofenac.5
Conclusions
In this randomised, blinded placebo-controlled study, IM diclofenac injection was associated with more advantage in time to pain reduction or overall efficacy as compared with PO administration. The findings were similar for the primary endpoint of substantial pain relief (ie, 50% drop in pain) and for a secondary endpoint of clinically significant pain reduction (NRS drop of 2). Given the small differences found between the two routes, and additional resources entailed in IM administration, this study suggests that when clinical circumstances allow a choice in NSAID administration route, the PO route is preferable.
References
Footnotes
Contributors IQ contributed by helping in the conception/design of the manuscript and critical review of the manuscript.SAP helped in formulating the research question. He also contributed in forming the statement of hypothesis and drafting of the initial manuscript. He also aided in the critical revision of the article. RSQ helped in data collection and helped in formulating the research question. SHT contributed in forming the concept and design of the manuscript; helped in data analysis and its interpretation; provided the final approval for the manuscript to be sent in for publication.
Funding The study was funded by Hamad Medical Corporation, Medical Research Center, Doha, Qatar (MRC#16413/16).
Competing interests None declared.
Ethics approval The trial was approved by the Hamad General Hospital Institutional Ethics Board, Doha, Qatar. Trial registrationnumber NCT03472339.
Provenance and peer review Not commissioned; externally peer reviewed.
Patient consent for publication Not required.