Article Text

Abstract
Objective To understand more about the individual variation in the time course of fibrinolysis following major injury and to assess the potential for stratification of trauma patients for tranexamic acid (TXA) therapy.
Methods A historical dataset (from 2004) was used, consisting of samples from 52 injured patients attended by a medical prehospital system. Blood samples were taken at the incident scene, on arrival in the emergency department, 2.5 hours after hospital arrival and 5 hours after hospital arrival. From the study database, we extracted values for tissue-type plasminogen activator (tPA; an activator of fibrinolysis), one of the plasminogen activator inhibitors (PAI-1; as a natural inhibitor of fibrinolysis) and D-dimer (as a marker of the extent of fibrinolysis).
Results The changes over time in median tPA and PAI-1 were mirror images, with initial high tPA levels which then rapidly decreased and low initial PAI-1 levels which slowly increased. There were high levels of fibrinolytic activity (D-dimer) throughout. This pattern was present in patients across a broad range of injury severities.
Conclusions After major trauma, there seems to be an early ‘antifibrinolytic gap’ with the natural antifibrinolytic system lagging several hours behind the natural profibrinolytics. An early dose of exogenous antifibrinolytic (TXA) might have its effect by filling this gap. The finding that tPA and subsequent clot breakdown (illustrated by D-dimer formation) are raised in a broad range of patients, with little correlation between the initial fibrinolytic response and markers of injury severity, may be the reason that TXA is effective across a broad range of injured patients.
- trauma
- major trauma management
- haematology
Statistics from Altmetric.com
Key messages
What is already known on this subject?
Tranexamic acid significantly improves survival after severe injury.
Early treatment with tranexamic acid is more effective.
In animals, there is an activation of fibrinolysis immediately after the injury.
What this study adds?
Profibrinolytic activity occurs within minutes after injury in humans, in contrast natural antifibrinolytic activity gradually increases over a number of hours.
We hypothesise that there is an ‘antifibrinolytic gap’ of unopposed fibrinolytic activity immediately after the injury.
There is an unexplained variation between patients.
Introduction
Since the publication of the CRASH2 trial results1 in 2010, an early dose of tranexamic acid (TXA) has been incorporated into major trauma management protocols worldwide.2 There is good evidence that TXA is most effective if given as early as possible (within the first hour) after injury.3 4 However, there has been some criticism of the universal use of this treatment and discussion about whether only selected patients should be treated.5 In addition, in many hospitals, TXA has been included in the massive haemorrhage protocol, with the implication that it will be given only to the most severely injured.6 This is not in line with the CRASH2 results, as in the trial it was given to a much wider group—all patients who had severe bleeding or had the potential for severe bleeding. A better understanding of the early mechanism of coagulation after injury could inform this debate.7
The most popular model of coagulation following injury includes four phases8 (initiation, amplification and propagation then fibrinolysis). Immediately after vascular injury, haemostatic thrombosis takes place with tissue factor, platelets and fibrinogen playing essential roles at this stage. Simultaneously, the release of the coagulation inhibitors, such as activated protein C and antithrombin, prevents the overspread of coagulation. Although the process is described in a series of stages, in fact, as a part of balanced coagulation, both coagulation inhibition and fibrinolysis start to occur as soon as coagulation begins (figure 1). So, the model of four sequential phases of coagulation is an oversimplification that has been constructed to represent the overall balance between multiple underlying promotor and inhibitory processes.

Dynamic balance of clot formation and breakdown after injury.
In the fibrinolytic system, the activation of plasminogen by tPA results in the early formation of fibrin degradation products, such as D-dimer, which further promotes fibrinolysis. Eventually, PAI-1, plasmin inhibitors (eg, α2-antiplasmin) and hepatic clearance of plasminogen activators work to regulate fibrinolysis and to curb the occurrence of overfibrinolysis in most patients.9 Fibrinolytic activity is increased in many trauma patients and is poorly detected by thromboelastography (TEG).10 Like all of the processes involved in clot formation and breakdown, fibrinolysis has a complex ‘yin and yang’ of promoting and inhibiting factors that maintain a balance. A lack of balance can lead to inappropriately low or high activity, and number of different pathological fibrinolytic syndromes have been postulated.11
TXA is a synthetic derivative of the amino acid lysine and has a very high affinity for the lysine-binding sites of plasminogen. Once these sites are occupied by TXA binding of plasminogen to fibrin is inhibited, which prevents activation by tPA thus creating an antifibrinolytic effect.9 We have previously shown that activation of both clot formation and clot breakdown happens very early after injury (within minutes).12 Functional studies have shown that early TXA is associated with increased clot strength, avoidance of hyperfibrinolysis and reduced coagulopathy.13 However, the variation between individuals and the relationship to injury severity has not been previously investigated. If all patients have early active fibrinolysis after injury, then this would be additional evidence that all patients should be treated with an antifibrinolytic; however, if there are individuals with and individuals without active early fibrinolysis, then a selective policy could be tested. In theory, patients might be stratified by the severity of their injury or according to the ratios between the promoters and inhibitors of fibrinolysis. However, a ‘precision medicine’ approach to patient stratification for TXA therapy is not currently possible as there is little published about the individual variation in the pattern of change of profibrinolytic and antifibrinolytic molecules.
We realised that some of the recent questions about the fibrinolytic system in injured patients might be approached by a reanalysis of an existing dataset of coagulation factors following injury (collected in 2004). The aim of our reanalysis was to define the pattern of change and individual variation in the time course of fibrinolysis following major injury and to assess the potential for stratification of trauma patients for TXA therapy.
Methods
This study was carried out by analysis of part of a dataset from 2004, which had been collected as part of an MD thesis, and examined both coagulation and fibrinolysis after severe injury.12 A grant for the original study was received from the Special Trustees of Barts and the Royal London Hospitals. The data were collected from a convenience sample of 52 injured patients attended by a medical prehospital system (sample determined by when the lift capacity of the helicopter was sufficient to carry a researcher in addition to the clinical team). Permission for the study was obtained from East London and The City Research Ethics Committee using the approvals system that was current at the time, and individual consent from patients/relatives was obtained.
The first venous blood sample was taken as soon as possible after injury at the time of placement of a cannula by the prehospital medical team (about 20 min after injury). Subsequent samples were taken on arrival in the emergency department (ED) (median 1 hour after injury), and then at 2.5 and 5 hours after injury. The prehospital samples were collected into citrate, EDTA and plain tubes and then packed in a cold (ice cooled) container for transfer. All samples were centrifuged on arrival in the ED and plasma was immediately frozen to –80°C. Samples remained frozen until analysis, which took place in two batches within 3 months of injury. Patient age, gender and outcome (lived or died) were recorded. Injury severity score (ISS) was calculated by a trained major trauma audit coder using the conventional scoring system.
From the study database, we extracted values for tissue-type plasminogen activator (tPA; an activator of fibrinolysis), one of the plasminogen activator inhibitors (PAI-1; as a natural inhibitor of fibrinolysis) and D-dimer (as a marker of the extent of fibrinolysis). Patients with missing data for a specific variable were excluded from the reporting of that variable. Values that were reported in words as below a detection threshold (such as ‘<0.01’) were assigned a numerical value at the detection threshold (ie, ‘0.01’) for the analysis. The median and IQR for each of these parameters was calculated at each time point (prehospital, ED, 2.5 and 5 hours) and plotted to show the relative changes. The individual patient data were plotted at each time point to illustrate the individual variation within these datasets.
In clinical care, it would, in theory, be possible to stratify patients by their provisional injury severity (calculated soon after arrival in hospital), so the ISS14 for each patient was extracted from the database and correlations between each parameter and injury severity were calculated using a Spearman rank correlation.
As we expected that a large initial fibrinolytic response would be followed by a large natural antifibrinolytic response, the correlation between the peak level of tPA and the peak level of PAI-1 was calculated using a Spearman rank correlation.
Statistical analysis was performed using SPSS V.24, and PRISM (GraphPad Software 7 Inc). Due to significant skew, the data were transformed using a two-step approach15 to a normal distribution (tested using the Shapiro-Wilk normality test). The significance of the overall pattern of change was then analysed on the transformed data using a one-way analysis of variance (ANOVA) with Tukey multiple comparison testing. A p value of<0.05 was taken to represent statistical significance.
Patient and public involvement
There was no patient or public involvement in the work.
Results
Fifty-two major trauma patients were included in the database. Of these, six patients (12%) died. All injuries were caused by blunt trauma. The median age was 45 (IQR, 27–61), with 69% being men. The median ISS was 31 (IQR, 19–39), with 87% of patients having an ISS of ≥16. The median prehospital fluid infused was 500 mL (IQR, 225–1200), the median ED fluid infused was 1500 mL (IQR, 500–2500 mL).
The summary of changes in the parameters measured to monitor the fibrinolytic system in the prehospital and early in-hospital phases of care are shown in table 1. There were more missing data for the D-dimer measurements than for tPA or PAI-1.
Changes in biomarkers associated with fibrinolysis (median and IQR)
The changes over time in median tPA and PAI-1 were mirror images, with initial high tPA levels which then rapidly decreased (figure 2) and low initial PAI-1 levels which slowly increased. There were high levels of fibrinolytic activity (D-dimer) throughout.

Patterns of change in median values of TPA, PAI-1 and D-dimer. ED, emergency department; tPA, PAI-1, plasminogen activator inhibitor 1; tPA, tissue plasminogen activator.
The distribution of changes in individual patients is shown in figures 3–5. For tPA (figure 3), levels were much higher in the prehospital phase, were already reducing on arrival in hospital and in most patients had returned to near zero by 2.5 hours after the injury (one-way ANOVA p<0.0001).
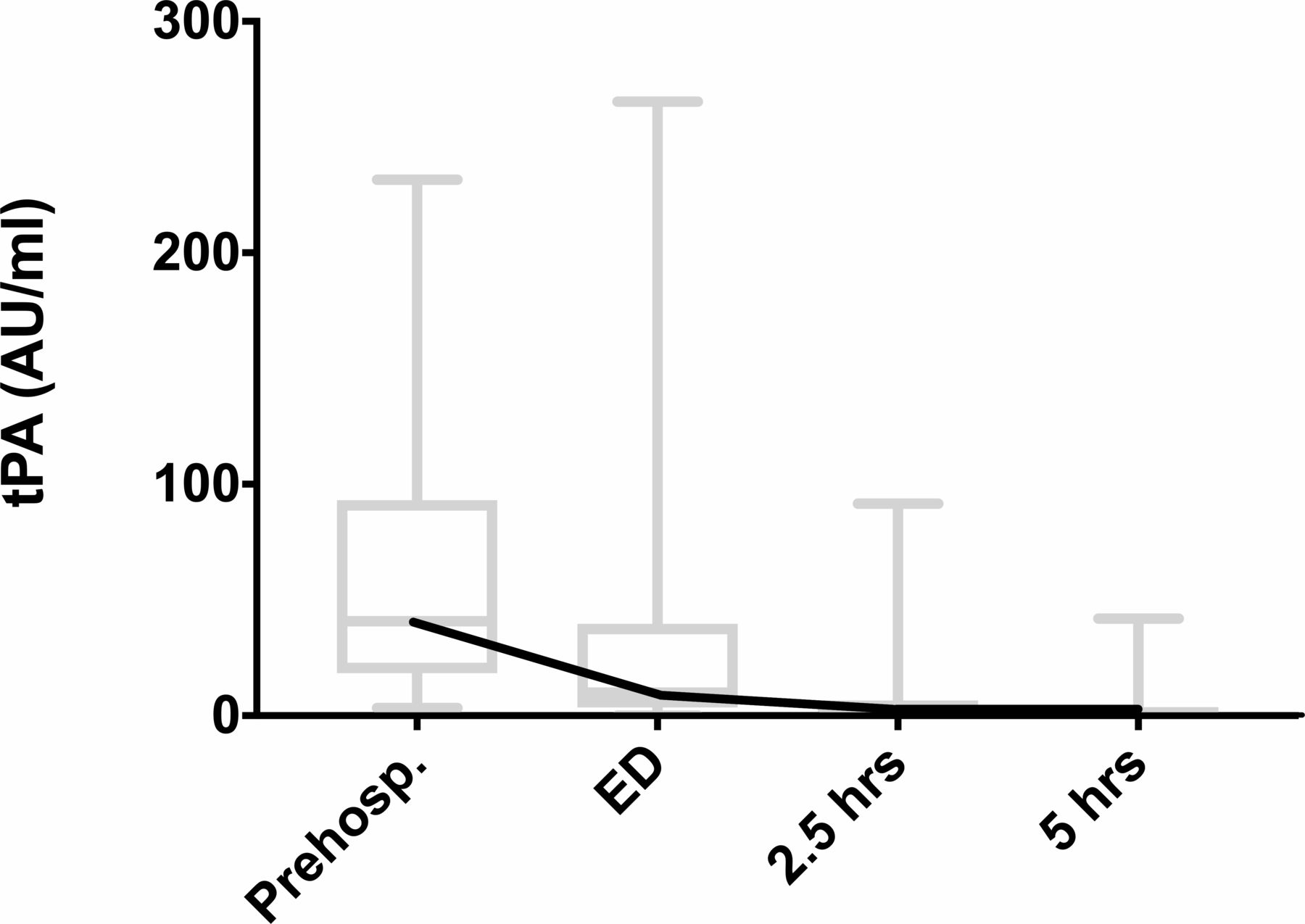
Individual variation of tPA levels. ED, emergency department; tPA, tissue plasminogen activator.

Individual variation in PAI-1 levels. ED, emergency department; PAI-1, plasminogen activator inhibitor 1.

Individual variation in D-dimer levels. ED, emergency department.
For PAI-1 (figure 4), levels were mainly low in the prehospital samples, had started to increase by ED arrival and were high by 2.5 hours, remaining high at 5 hours (one-way ANOVA p<0.0001).
D-dimer levels were above normal in the prehospital samples and remained high (figure 5), with a trend towards a decrease at 5 hours (Tukey multiple comparison test, ED vs 5 hours p=0.045).
There was little or no correlation between ISS and the markers of fibrinolysis and antifibrinolysis (table 2). The weak correlation between peak tPA and ISS is shown in figure 6.
Correlation between biomarkers of fibrinolytic activity and ISS
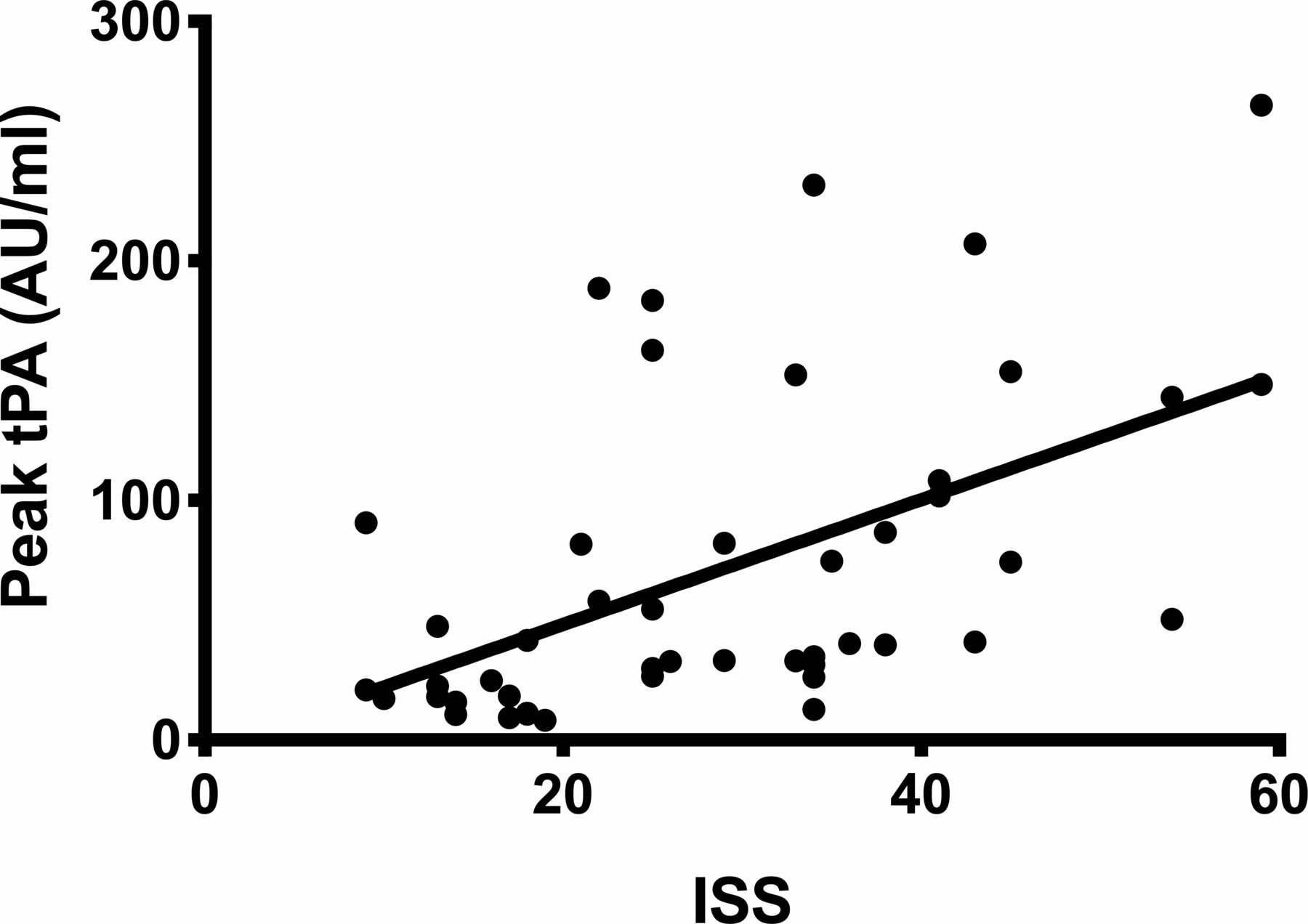
Correlation between peak tPA and ISS (r=0.57). ISS, Injury Severity Score; tPA, tissue plasminogen activator.
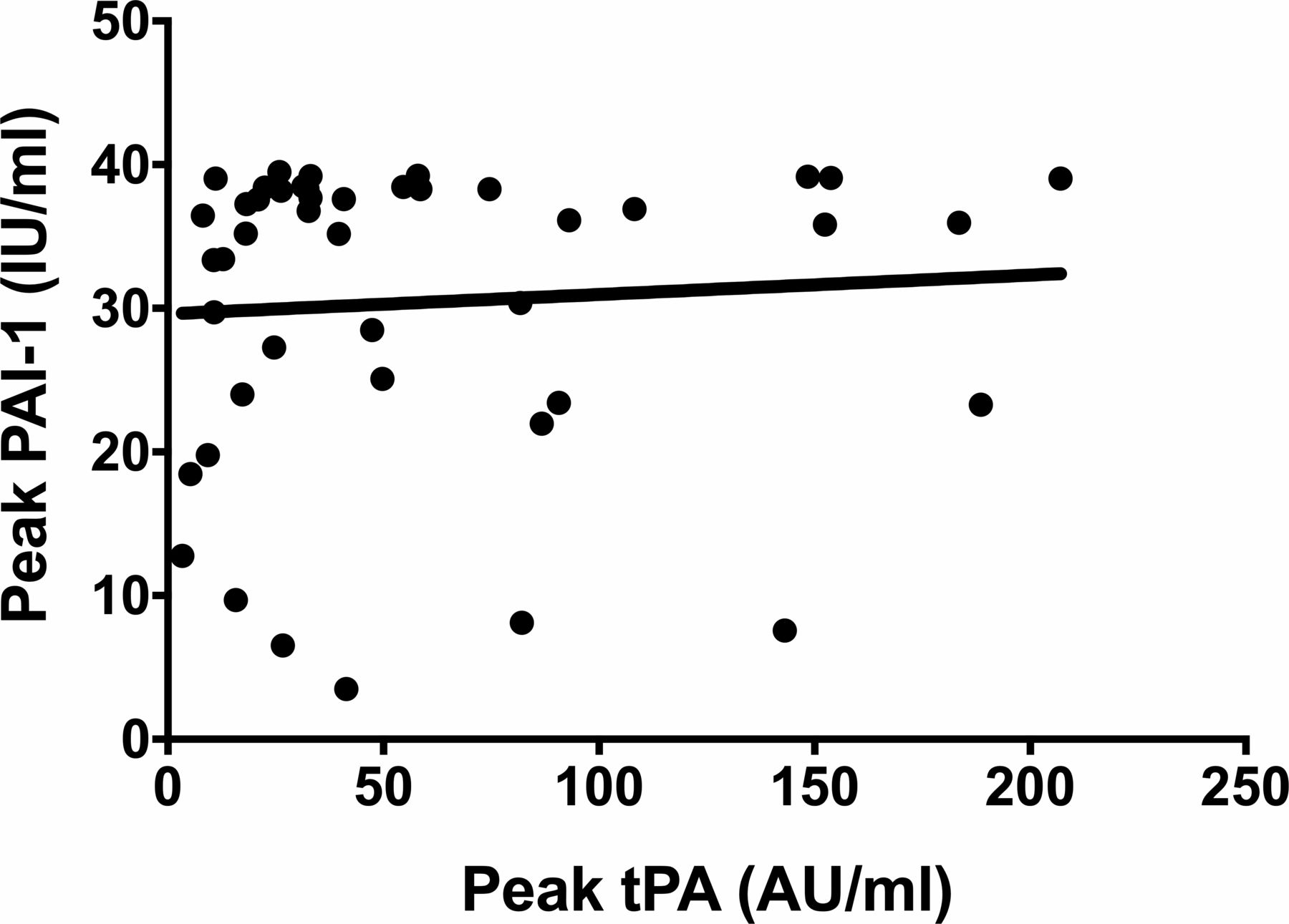
The was no correlation (r=0.16) between the peak level of tPA (the prehospital value) and the peak level of PAI-1 (the 5-hour value) (figure 7).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Correlation between the peaks of tPA and PAI-1 (r=0.16). PAI-1, plasminogen activator inhibitor 1; tPA, tissue plasminogen activator.
Discussion
We have previously reported the overall changes (table 1 and figure 1) which show that this group of patients has a very early peak of tPA and D-dimer, indicating a maximum of fibrinolytic activity at about the time of arrival in the ED.12 The current analysis looks at the distribution of individual values for the three key markers of profibrinolytic, fibrinolytic and antifibrinolytic activities.
When the distribution of individual values of the pro-fibrinolytic (tPA) was examined, all patients were found to have a raised tPA on the initial measurement in broad distribution. However, reflecting its short half-life, many patient’s levels had reduced to near zero by the time the patient arrived in the ED (at about an hour after injury). By 5 hours after an injury nearly all patients had returned to very low levels of tPA activity.
The distribution of individual values of the natural antifibrinolytic (PAI-1) showed a different temporal pattern of change, with early low levels followed by a rise to the highest levels at 5 hours (as measurement stopped at 5 hours the peak in PAI-1 levels may be later than this and been missed by this study). This lag in production of the natural antifibrinolytic gave an ‘antifibrinolytic gap’ in the first few hours after injury, during which the profibrinolytic effect of tPA seemed to be unopposed.
The end effect of the balance between pro and antifibrinolytic influences (the extent of clot breakdown) is shown in the D-dimer measurements, with a peak occurring at around the time of arrival in the ED. This could also reflect early unopposed profibrinolytic activity before the natural antifibrinolytic effect of PAI-1 is activated. We hypothesise that the explanation for unopposed fibrinolysis may be in the different mechanisms of synthesis and release of the profibrinolytic and antifibrinolytic molecules. Profibrinolytic tPA is rapidly released as it is already synthesised and stored in secretory vesicles within the endothelium,16 17 in contrast, production of the antifibrinolytic PAI-1 needs the cellular signalling system to operate and PAI-1 mRNA to become translationally active to then produce the antifibrinolytic molecule,18 causing a time lag.
Our data show that there is little or no correlation between ISS and initial measurements of coagulation parameters associated with fibrinolysis (tPA, PAI-1 and D-Dimer). This is a different conclusion from previous studies carried out using later measurements from intensive care unit patients19 (maybe due to earlier measurement), and suggests that it is not possible to use injury severity to stratify the early fibrinolytic response. This implies that factors other than just injury severity are important in determining the fibrinolytic response. Potential other factors are; genetic variations, variable immune response, differences in fibrinolytic between injury types with the same ISS, and the overall degree of microvascular endothelial damage (which may be poorly related to ISS). The lack of correlation of injury severity and fibrinolytic response is consistent with the clinical evidence from CRASH2. Treatment with an antifibrinolytic was found to be effective over a broad range of injury severity, with low risk patients also benefiting.20
The lack of correlation between ISS and activity of the fibrinolytic system also has an implication for trauma care research, as coagulation studies that use ISS to adjust for differences between groups5 are likely to be open to bias. The current lack of knowledge about the basic biological mechanisms of clot formation and breakdown after severe injury makes it difficult to confidently interpret observational data.
The coagulation system has evolved an exquisite and complex system of checks and balances that maintains blood as a fluid but converts just the right amount to a solid clot when necessary. So, it seems surprising that there is an ‘antifibrinolytic gap’ of unopposed fibrinolysis. One hypothesis may be that the human coagulation system has not had an opportunity to evolve an appropriate response to major trauma, as in human history it is only relatively recently that those suffering major trauma have survived to pass on their genes. There has therefore been no genetic selection pressure to create an appropriate response to major trauma. An alternative explanation for the apparent ‘antifibrinolytic gap’ is that we simply do not understand all of the complex influences fibrinolysis after severe injury, as there is a complex series of feedback loops and cross-talk between inflammatory and coagulation systems.
The genetic influences on the natural history, biology and outcome following major trauma have been little researched. However, by extrapolation from other diseases it is likely that currently unknown genetic factors play a major role in determining the changes in blood clotting and clot breakdown after injury. This is likely to account for some of the variation between individuals that we have observed and seems to be a key area for future trauma care research.
There seems to be a synergy between the molecular and clinical data about TXA treatment. Both suggest that TXA treatment should be regarded as a ‘primary survey’ critical intervention and should not be included in a ‘massive transfusion protocol’, as it should have been given before this stage of patient care is reached. Exogenous antifibrinolytic (TXA) needs to be given soon after injury (in the prehospital phase of care) if it is to fill the ‘antifibrinolytic gap’ that has been demonstrated before the natural (intrinsic) antifibrinolytic response. This fits very well with the clinical observation of much more benefit from early treatment.4
Our study has a number of limitations. It is based on historical data from a time before TEG became widely available; however, we would argue that TEG is not a relevant test in TXA treatment as there is too long a delay to get the results from the fibrinolytic phase for it to be used to stratify patients (our data emphasise the need for very early treatment) and is insensitive to all except extreme changes in fibrinolysis.10 In our dataset antiplasmin (another natural antifibrinolytic substance) was not measured due to analytical constraints. There are no data about the time course of antiplasmin activity in major trauma; however, it has to be synthesised within cells (like PAI-1), and in other conditions, it rises over days rather than hours.21 This study was carried out in a small sample and there were no patients with penetrating trauma (reflecting the normal UK major trauma population in 2004) so our results can only be definitely applied to blunt trauma. Details of the precise laboratory methods used in 2004 were not preserved with the data (a lesson for those preserving research datasets). This makes interpretation of absolute levels difficult, however our conclusions depend on patterns of change over time rather than absolute values. The most significant limitation may be that we are only able to measure levels of molecules in the general circulation. Circulating molecules are the ‘overspill’ from local processes, are affected by both the rate of overspill and the rate of clearance, and so may be very different from the local concentrations in the damaged blood vessels. There were also missing data for D-dimer in about half of patients, however it is well known that D-dimer is raised even after relatively small injuries, so these missing data are unlikely to affect the conclusions of this paper or the proposed hypothesis.
Conclusions
Analysis of the fibrinolytic system in major trauma may have provided a biological explanation for the clinical finding that patients across a broad range of probabilities of survival benefit from TXA therapy and that early treatment have much more benefit.13 14 After major trauma, there seems to be an early ‘antifibrinolytic gap’ with the natural antifibrinolytic system lagging several hours behind the natural profibrinolytics. An early dose of exogenous antifibrinolytic (TXA) might have its effect by filling this gap. The finding that tPA and subsequent clot breakdown (illustrated by D-dimer formation) are raised in a broad range of patients with little correlation between the initial fibrinolytic response and markers of injury severity is consistent with the clinical evidence that TXA should not be restricted to the most seriously injured patients.
Our reanalysis of an old dataset is hypothesis forming rather than definitive. Further studies using modern analytical methods (and maybe also genetic analysis) are urgently required to either prove or disprove our theory of an ‘antifibrinolytic gap’ and to better understand the variation in response between individuals.
References
Footnotes
Twitter @TJCoats
Contributors Both authors were involved in each stage of the development of the idea, extraction and analysis of the data and writing of the manuscript.
Funding The original data collection was funded by the Special Trustees of Barts and the Royal London Hospital with ethics approval from the East London and The City Research Ethics Committee.
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request. Data are available through the corresponding author.